



RBM8A promotes gastric cancer progression by binding with UPF3B to induce BBC3 mRNA degradation
- Authors:
- Published online on: June 30, 2025 https://doi.org/10.3892/ijmm.2025.5572
- Article Number: 131
-
Copyright: © Peng et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Gastric cancer (GC), a malignant tumor of the digestive tract, is the fifth most prevalent tumor and the fourth most lethal tumor globally (1). The principal treatment modalities for GC encompass surgery, chemotherapy, targeted therapy and immunotherapy. However, due to the high heterogeneity of GC in terms of phenotype and genetics, some patients with GC are not sensitive to various anticancer treatments (2). The overall treatment effectiveness and prognosis are still unsatisfactory (2,3). Therefore, it is of great significance to explore the potential key regulators of GC and to elucidate their biological functions and potential molecular mechanisms in order to identify molecular targets of drugs.
The RNA binding motif protein 8A (RBM8A) gene encodes the protein Y14, which contains a conserved RNA-binding motif. This protein consists of three domains: An N-terminal localization signal, a C-terminal short arginine/serine repeat sequence region and a central RNA recognition motif involved in MAGOH binding (4). RBM8A is distributed in both the cytoplasm and the nucleus, and shuttles between these compartments (5). RBM8A forms a heterodimer with MAGOH. Together with eukaryotic initiation factor 4A-III and CASC3, it constitutes the exon-junction complex (EJC), which is involved in RNA splicing, subcellular localization and nonsense-mediated mRNA decay (NMD) processes (6). As one of the core proteins of the EJC, RBM8A participates in most stages of the mRNA life cycle and holds an important regulatory position in each stage of gene expression due to its impact on mRNA metabolism (7). Studies have shown that when there is a premature stop codon, as the ribosome reaches the stop codon, the EJC remains downstream of the ribosome and sends a signal to the ribosome indicating that the stop codon appears prematurely. Subsequently, the ribosome recruits the SMG-1-Upf1-eRF1-eRF3 complex to interact with the EJC and marks the mRNA through UPF1-mediated phosphorylation, leading to mRNA degradation (7-11). Some mRNAs that lack premature stop codons but have special features, such as long 3′ untranslated regions (UTRs), exon-exon junctions with >50 nt downstream of the stop codon, long upstream open reading frames and introns in the 3′UTR, are also marked and degraded (8,12-14). This indicates that RBM8A serves a crucial role in mediating mRNA metabolism.
A previous study has demonstrated that RBM8A is upregulated in various tumors, including breast cancer, bladder cancer, cervical cancer, colorectal cancer, head and neck squamous cell carcinoma, liver cancer, and glioma (15). This indicates the universality of RBM8A upregulation during cancer progression. Studies have found that the upregulation of RBM8A in glioma, breast cancer and liver cancer promotes the proliferation, migration and invasion of cancer cells, and inhibits their apoptosis, suggesting that RBM8A serves an important role in cell survival (16-18). RBM8A is also involved in the regulation of cisplatin resistance in breast cancer cells (19). The aforementioned studies have revealed that RBM8A is involved in the regulation of several cellular processes associated with cancer development, including apoptosis. However, to the best of our knowledge, currently, there is no research on the biological functions of RBM8A in GC cells. BCL2 binding component 3 (BBC3), a key pro-apoptotic protein, was investigated as a target in the present study because its dysregulation has been linked to the evasion of apoptosis in GC cells (20), which is a crucial step in tumorigenesis and cancer progression.
The present study investigated the biological functions and potential molecular mechanisms of RBM8A in GC progression. The present study explored the biological roles of RBM8A through in vitro functional assays and in vivo nude mouse xenograft tumor experiments, and systematically screened and validated downstream target genes regulated by RBM8A using mRNA-sequencing (mRNA-seq) and RNA immunoprecipitation (RIP)-seq. Furthermore, actinomycin D (Act D) assays and co-immunoprecipitation (CoIP)-mass spectrometry (MS) were performed to elucidate the underlying molecular mechanisms by which RBM8A modulated target gene expression levels to promote GC progression.
Materials and methods
Bioinformatics analysis
Gene expression data were obtained from The Cancer Genome Atlas (TCGA; https://xena.ucsc.edu) and Genotype-Tissue Expression (https://gtexportal.org/) public databases. RBM8A expression in GC and the survival curve of patients with GC based on RBM8A expression were analyzed using the Xiantao website (https://www.xiantaozi.com/). For the Kaplan-Meier survival analysis, the present study utilized the TCGA GC mRNA dataset and focused on the 5-year survival rate for all subjects to minimize the impact of late-stage crossover on the test results. Subjects were divided into RBM8A high expression and low expression groups based on the median expression level of RBM8A. The log-rank test was used to compare the survival distributions between the groups within this 5-year window.
Tissue microarray and multiplex immunohistochemistry (mIHC)
A GC tissue microarray (cat. no. D160St01) containing 80 pairs of cancerous tissues and adjacent non-cancerous tissues was purchased from Zhongke Guanghua (Xi'an) Intelligent Biotechnology Co., Ltd. The tissues were fixed with 10% neutral-buffered formalin at room temperature for 24 h prior to processing. The present study was reviewed and approved by the Medical Ethics Committee of Shaanxi Provincial People's Hospital (approval no. 2023-R176; Xi'an, China). The mIHC assay was performed using an AlphaXTSA Multiplex Immunohistochemistry kit [cat. no. AXT37100021; Aikefa (Beijing) Biotechnology Co., Ltd.] according to the manufacturer's protocols. Briefly, the paraffin-embedded sections (4 μm thick) were incubated at 59°C for 1 h, deparaffinized in xylene (2×10 min), rehydrated using a descending ethanol series (100, 95, 80 and 70%) for 5 min each, and washed in distilled water. Antigen retrieval was performed by boiling sections in 10 mM sodium citrate buffer (pH 6.0) for 15 min using a pressure cooker. Sections were then permeabilized with 0.3% Triton X-100 in PBS for 15 min at room temperature, and endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 15 min at room temperature. Slides were washed three times with PBS (pH 7.4) for 5 min each, and blocking was carried out using 5% BSA (GC305010; Wuhan Servicebio Technology Co., Ltd.) in PBS for 1 h at room temperature. The sections were incubated with primary antibodies [anti-RBM8A, 1:500, cat. no. ab181038, Abcam; anti-pan-cytokeratin (PANCK), 1:500, cat. no. ZM-0069, Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.] at 37°C for 1 h. The sections were then washed three times with TBS with 0.1% Tween-20 (TBST). The sections were incubated with anti-rabbit/mouse secondary antibodies from the AlphaXTSA kit (cat. no. AXT37100021) at a 1:3 dilution at 37°C for 10 min. After washing three times with TBST, fluorescent dye from the AlphaXTSA kit (cat. no. AXT37100021) diluted in signal amplification solution (1:100 dilution) was used for staining at room temperature for 5 min. Antigen retrieval was repeated using sodium citrate retrieval solution in a microwave oven at low power for 15 min, followed by cooling to room temperature. The steps from blocking to antigen retrieval were repeated until staining with the last antibody was completed. After DAPI staining at room temperature for 5 min and washing with TBST three times, the sections were mounted with an anti-fluorescence quenching agent for imaging analysis. Imaging was performed using a fluorescence microscope (Axioscan; Zeiss GmbH). For analysis, the fluorescence intensity of each channel was quantified using Halo software (version 4.0; Indica Labs, Inc.).
Cell culture
AGS, HGC27 and MKN45 GC cell lines authenticated by STR were obtained from the following sources: AGS and MKN45 cells were purchased from Procell Life Science & Technology Co., Ltd., while HGC27 cells were obtained from Beyotime Institute of Biotechnology. The cell lines were subsequently maintained at the Biomedical Experimental Center of Xi'an Jiaotong University (Xi'an, China). The cells were cultured in RPMI1640 (Cellmax) supplemented with 10% FBS (Sangon Biotech Co., Ltd.) and 1% penicillin-streptomycin solution (Sangon Biotech Co., Ltd.). 293T cells obtained from the Biomedical Experimental Center of Xi'an Jiaotong University (Xi'an, China; originally from American Type Culture Collection) were cultured in DMEM (Cellmax) supplemented with 10% FBS and 1% penicillin-streptomycin solution. All cells were incubated and maintained at 37°C in a humidified incubator with an atmosphere containing 5% CO2.
Transfection of small interfering RNA (siRNA/si) and plasmid
For knockdown, two siRNAs for RBM8A were designed to minimize off-target effects, and a siRNA for BBC3 was also designed. siRNA targeting the dual luciferase gene was utilized as a negative control (siNC). All siRNAs were purchased from Sangon Biotech Co., Ltd. The efficiency of RBM8A knockdown was verified using reverse transcription-quantitative PCR (RT-qPCR) and western blotting. The coding DNA sequence regions of RBM8A and UPF3B were cloned into the pLV3 plasmid vector (P68154; MiaoLingBio) for overexpression. The empty pLV3 vector was used as the negative control for overexpression experiments. The shRBM8A primer, which has a common target sequence with siRBM8A-1, was annealed and cloned into the pLKO.1 plasmid vector (V010450; NovoPro). The sequences of siRNAs and plasmid cloning primers are listed in Table I. Transfection of siRNAs and plasmids was performed using Lipofectamine 2000 reagent (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. In 6-well plates, for siRNA transfection, 5 μl of 100 μM siRNA stock solution (final concentration, 50 nM) was used per well. For plasmid transfection, 2 μg of plasmid DNA was added to each well. The transfection complexes were incubated at room temperature for 10 min, then added to cells in Opti-MEM reduced serum medium (31985070; Thermo Fisher Scientific, Inc.). Cells were cultured in a 37°C incubator with 5% CO2 for 6 h, after which the medium was replaced with complete growth medium. Subsequent experiments were performed 48 h after transfection.
RNA isolation and RT-qPCR
Total RNA was extracted from of GC cells or tissues using AG RNAex Pro Reagent (AG21101; Hunan Accurate Bio-Medical Technology Co., Ltd.), followed by reversing transcription using an Evo M-MLV reverse transcription kit (Hunan Accurate Bio-Medical Technology Co., Ltd.) at 37°C for 15 min. qPCR was performed using SYBR Green Pro Taq HS (Hunan Accurate Bio-Medical Technology Co., Ltd.) on a Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.) with the following thermocycling conditions: 95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30 sec. Relative expression levels of genes were quantified using the 2−ΔΔCq method (21). GAPDH was used as an internal reference for quantification purposes. All the specific primer sequences used in the present study are listed in Table I.
Cell Counting Kit-8 (CCK8) assay
Cells transfected with siNC or siRNA targeting RBM8A or BBC3 were seeded into 96-well plates (3×103 cells per well), and then the plates were placed into a 37°C incubator with 5% CO2. After 24, 48 or 72 h, 10 μl CCK8 reagent (NCM Biotech) was added and cells were incubated for 2 h. The absorbance at 450 nM was detected with a microplate reader.
Colony formation assay
A total of 1×103 cells per well transfected with siNC or siRNA targeting RBM8A or BBC3 were seeded into 6-well plates and cultured for 7-10 days in an incubator at 37°C with 5% CO2. The cells were fixed with 4% paraformaldehyde for 30 min at room temperature. The cells were then washed three times using PBS, followed by staining with 0.1% crystal violet for 30 min at room temperature. Colonies were quantified manually by counting with an ordinary optical microscope (Olympus SZX10; Olympus Corporation), and colonies containing >50 cells were counted.
Flow cytometry
To assess apoptosis, cells transfected with siNC or siRNA targeting RBM8A or BBC3 were harvested and then double stained with FITC-conjugated annexin V and PI (Annexin V-FITC/PI Apoptosis Detection kit; Beyotime Institute of Biotechnology). The percentage of early and late apoptotic cells was analyzed using flow cytometry (ACEA NovoCyte 1040; ACEA Biosciences, Inc.) with ACEA NovoExpress software (version 1.5.0; ACEA Biosciences, Inc.) for data acquisition and analysis.
Cell migration assay
Cells transfected with siNC or siRNA targeting RBM8A were seeded in a 6-well plate. After the confluence rate reached 100%, a scratch was created using a 200-μl pipette tip. The cells were washed with PBS three times to remove the cells scraped off by the pipette tip, serum-free medium was added and the cells were cultured in the incubator. Images of the cells were captured using an inverted fluorescence microscope (DMi8; Leica Microsystems GmbH) with bright-field imaging at 0, 24 and 48 h after scratching.
CoIP assay and protein MS
Cells (AGS or HGC27) were transfected with RBM8A-3 x Flag or UPF3B-3 x Flag overexpression plasmids (pLV3 backbone; P68154; MiaoLingBio) using Lipofectamine 2000 (cat. no. L3000015; Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. After 48 h of transfection, cells were lysed using RIPA Lysis Buffer (P0013D; Beyotime Institute of Biotechnology) supplemented with protease inhibitors, and the lysates were centrifuged at 12,000 × g for 10 min at 4°C. The supernatant (500 μg of total protein per IP reaction) was collected. The following primary antibodies (2 μg each) were added to the lysates: Anti-Flag (cat. no. 20543-1-AP; Proteintech Group, Inc.) for Flag-tagged proteins or anti-rabbit IgG (cat. no. 30000-0-AP; Proteintech Group, Inc.) as a negative control. The samples were mixed overnight at 4°C with slow rotation. Protein A/G Magnetic Beads (40 μl) were added and samples were mixed for 1 h at 4°C with slow rotation. After magnetic separation, the samples were washed five times with wash buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40) by resuspending beads in buffer and separating them using a magnetic stand. Proteins were eluted by adding protein loading buffer and incubation at 100°C for 5 min. Specific target proteins were detected by western blotting as described subsequently.
MS was performed by Shanghai GeneChem Co., Ltd. Protein samples obtained from immunoprecipitation with anti-IgG antibody and anti-RBM8A antibody were separated by 10% PAGE. Subsequently, in-gel digestion with trypsin was carried out, enabling the cleavage of proteins into smaller peptide fragments. These peptides were then extracted using 60% acetonitrile (ACN)/0.1% trifluoroacetic acid with sonication for 15 min, lyophilized and reconstituted in 0.1% formic acid. Liquid chromatography-tandem MS was performed using a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Inc.) coupled with an Easy nLC 1200 system (Thermo Fisher Scientific, Inc.). Ionization was achieved via nanoelectrospray ionization in positive ion mode with a spray voltage of 2.0 kV. The capillary temperature was set to 275°C, the S-lens RF level was 50% and the nebuliser pressure was set at 2.0 psi. Peptides were separated on a C18-reversed phase analytical column (Acclaim PepMap RSLC, 50 μm × 15 cm; Thermo Fisher Scientific, Inc.) with a linear gradient of buffer A (0.1% formic acid in water) and buffer B (80% ACN/0.1% formic acid) at a flow rate of 300 nl/min. The gradient was: 6% B for 5 min, 6-8% B over 40 min, 28-38% B over 5 min, 38-100% B over 5 min and 100% B for 5 min. For targeted quantification of selected peptides, multiple reaction monitoring (MRM) transitions were optimized using Skyline software (version 21.1; https://www.skylinesoft.com/). MRM data were acquired with a cycle time of 300 msec and a scan range of 200-2,000 m/z. Subsequently, library identification analysis was performed by employing Proteome Discoverer 2.2 software (Thermo Fisher Scientific, Inc.) in combination with the Mascot 2.6 (Matrix Science, Inc.) search engine.
Western blotting
Total protein was exacted from cells using RIPA Lysis Buffer (cat. no. P0013B; Beyotime Institute of Biotechnology) supplemented with protease and phosphatase inhibitors. The protein concentration was determined using a BCA Protein Assay kit (cat. no. P0010S; Beyotime Institute of Biotechnology) according to the manufacturer's instructions. A total of 20 μg of protein per lane was separated by 10% SDS-PAGE, then transferred onto a PVDF membrane (MilliporeSigma). The membrane was blocked with 5% skimmed milk powder (GC310001; Wuhan Servicebio Technology Co., Ltd.) at room temperature for 1 h, and then incubated with primary antibodies against RBM8A (1:1,000; cat. no. ab181038; Abcam), GAPDH (1:2,000; cat. no. ab181602; Abcam), poly(ADP-ribose) polymerase 1 (PARP1; 1:3,000; cat. no. sc-8007; Santa Cruz Biotechnology, Inc.), cleaved PARP1 (1:5,000; cat. no. 60555-1-Ig; Proteintech Group, Inc.), BAX (1:1,000; cat. no. 2772; Cell Signaling Technology, Inc.), BBC3 (1:3,000; cat. no. 4976; Cell Signaling Technology, Inc.), UPF3B (1:5,000; cat. no. 23301-1-AP; Proteintech Group, Inc.) and Flag (1:3,000; cat. no. sc-51590; Santa Cruz Biotechnology, Inc.) at 4°C overnight. After washing with TBST five times, the membrane was incubated with HRP conjugated goat anti-rabbit IgG secondary antibody (cat. no. L3012; 1:10,000; Signalway Antibody LLC) or HRP conjugated goat anti-mouse IgG secondary antibody (cat. no. L3032; 1:10,000; Signalway Antibody LLC) at room temperature for 1 h. Finally, chemiluminescent signals were detected using BeyoECL Plus Luminescent Solution (P0018S; Beyotime Institute of Biotechnology), the bands were examined using a GelView 5000Plus Chemiluminescent gel imaging system (Guangzhou Biolight Biotechnology Co., Ltd.). Densitometric analysis was performed using ImageJ software (version 1.46r; National Institutes of Health).
mRNA-seq
For mRNA-seq, siNC and siRBM8A-1 were transfected into AGS cells for 48 h. Total RNA was extracted using an AG RNAex Pro Reagent kit (AG21101; Hunan Accurate Bio-Medical Technology Co., Ltd.). The mRNA-seq experimental procedures were performed by Shanghai GeneChem Co., Ltd. Briefly, RNA integrity was verified using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.), with RNA integrity number (RIN) values ≥8.0 considered acceptable. mRNA with poly(A) tails was enriched using Oligo (dT) magnetic beads (E7490; New England BioLabs, Inc.). Subsequently, the mRNA was randomly fragmented. DNA libraries were prepared using the TruSeq® Stranded mRNA Library Prep kit (RS-122-2101; Illumina, Inc.). Sequencing was performed on an Illumina HiSeq 4000 platform (Illumina, Inc.) using the Illumina HiSeq 4000 SBS kit (cat. no. FC-401-4002; Illumina, Inc.) with paired-end 150-bp reads. The final library concentration was quantified using qPCR with KAPA SYBR FAST qPCR Master Mix in the KAPA Library Quantification kit for Illumina Platforms (KK4824; Roche Diagnostics) and normalized to 2 nM for loading. The thermocycling conditions were as follows: 95°C for 3 min (initial denaturation), 40 cycles of 95°C for 10 sec and 60°C for 30 sec, followed by melting curve analysis from 65-95°C (0.5°C/sec increment). Reactions were carried out on a LightCycler® 480 System II (Roche Diagnostics). The kit included a Premixed Primer Mix targeting the universal Illumina adapter sequences: Forward primer, 5′-AATGATACGGCGACCACCGAGATCTACAC-3′ (Illumina P5 primer); and reverse primer, 5′-CAAGCAGAAGACGGCATACGAGAT-3′ (Illumina P7 primer). Data analysis included the following: Raw read trimming with Trimmomatic (version 0.39; http://www.usadellab.org/cms/?page=trimmomatic), alignment to the human genome (GRCh38) using HISAT2 (version 2.2.1; https://daehwankimlab.github.io/hisat2/), transcript assembly and quantification with StringTie (version 2.2.1; https://ccb.jhu.edu/software/stringtie/), and differential expression analysis using DESeq2 (version 1.36.0; https://bioconductor.org/packages/release/bioc/html/DESeq2.html).
RIP assay and RIP-seq
For the RIP assay, the RNA Immunoprecipitation kit (Bes5101; Guangzhou BersinBio Biotechnology Co., Ltd.) was used according to the manufacturer's instructions. No plasmid vectors were used in this RIP assay, as endogenous RBM8A protein was directly immunoprecipitated without overexpression. Briefly, the cells were collected and washed once with PBS, then 1.7 ml polysome lysis buffer (Bes5101; Guangzhou BersinBio Biotechnology Co., Ltd.) with protease inhibitor (Bes5101; Guangzhou BersinBio Biotechnology Co., Ltd.) and RNAase inhibitor (Bes5101; Guangzhou BersinBio Biotechnology Co., Ltd.) was added to the cell precipitate and cells were lysed for 20 min on ice. DNA was removed using DNAase (Bes5101; Guangzhou BersinBio Biotechnology Co., Ltd.). After centrifugation at 10,000 × g for 10 min in 4°C, 5% (1/20 volume) of the supernatant was retained as the input fraction. The remaining sample was divided equally into two portions, 0.8 ml per IP reaction, and 2 μg anti-RBM8A antibodies (ab181038; Abcam) were added to the samples for incubation at 4°C overnight with rotation, while corresponding anti-rabbit IgG antibodies (Bes5101; Guangzhou BersinBio Biotechnology Co., Ltd.) were used as a negative control. Protein A/G magnetic beads (20 μl; Bes5101; Guangzhou BersinBio Biotechnology Co., Ltd.) were added to each set of samples for incubation at 4°C for 1 h with rotation. The magnetic beads were separated from the supernatant using a magnetic stand, followed by washing five times using polysome washing buffer. AG RNAex Pro reagent was added to the samples and RNA was extracted according to the manufacturer's method.
The RIP-seq of the RNA derived from the RIP assay was performed by Sangon Biotech Co., Ltd. Briefly, RNA integrity was verified using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.) with RIN values ≥7.0 considered acceptable. Sequencing libraries were generated using the NEBNext Ultra II Directional RNA Library Prep kit for Illumina (E7760S/L; New England BioLabs, Inc.). Sequencing was performed on an Illumina HiSeq X Ten platform (Illumina, Inc.) using the HiSeq X Ten Reagent kit (cat. no. FC-501-2501; Illumina, Inc.) with paired-end 150-bp reads. The final library concentration was measured using qPCR with KAPA SYBR FAST qPCR Master Mix in the KAPA Library Quantification kit for Illumina Platforms (KK4824; Roche Diagnostics) under the following thermocycling conditions: 95°C for 3 min (initial denaturation), 40 cycles of 95°C for 10 sec and 60°C for 30 sec, followed by melting curve analysis from 65-95°C (0.5°C/sec increment) on a LightCycler® 480 System II (Roche Diagnostics) and diluted to 1.8 pM for clustering. The kit included a Premixed Primer Mix targeting the universal Illumina adapter sequences: Forward primer, 5′-AATGATACGGCGACCACCGAGATCTACAC-3′ (Illumina P5 primer); and reverse primer, 5′-CAAGCAGAAGACGGCATACGAGAT-3′ (Illumina P7 primer). Data analysis was performed using the following software: HISAT2 (2.1.0; https://daehwankimlab.github.io/hisat2/), SAMtools (1.15; https://www.htslib.org/doc/samtools.html) and MACS2 (2.2.7.1; https://github.com/macs3-project/MACS).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis
Differentially expressed genes (DEGs) were analyzed using the Database for Annotation, Visualization and Integrated Discovery Bioinformatics Resources 6.8 (https://davidbioinformatics.nih.gov/). Gene symbols of DEGs (adjusted P-value <0.05; |log2 fold change|>1) were uploaded as a text file with the entire genome of Homo sapiens as the background. GO enrichment analysis was performed for biological process, molecular function and cellular component categories with a modified Fisher's exact P-value (Enhanced Analysis of Structures Enrichment Score) ≤0.05 and minimum gene count of 2, followed by Benjamini-Hochberg (BH) correction for multiple testing. KEGG pathway analysis was conducted for Homo sapiens with a P-value ≤0.05 and false discovery rate adjustment using the BH method.
RNA pulldown assay
The RNA pulldown assay was performed as described previously (22). Briefly, biotin-labeled BBC3 mRNA-specific probe (5′-TCCATCAGCCGTCCCTCTCCTGGCTTC-3′-Biotin) and control probe (5′-AGATCACCAAGAGGTGCAACATTAG-3′-Biotin) were synthesized by Sangon Biotech Co., Ltd. Cells were fixed with 1% paraformaldehyde at room temperature for 15 min, and 1.25% glycine was utilized to terminate fixation. The cells were collected after washing with PBS, and lysed with 500 μl lysis buffer at 4°C, and sonicated for 30 sec followed by centrifuging at 12,000 × g at 4°C for 10 min, and the supernatants were collected for hybridization. BBC3 mRNA-specific probe or control probe (100 nM) was added for hybridization at 20°C overnight. Streptavidin-conjugated magnetic beads were added and samples were incubated with rotation at room temperature for an additional 2 h. Subsequently, the samples were de-crosslinked and magnetic beads were discarded, and the supernatants were collected for RNA isolation with AG RNAex Pro reagent or western blotting with protein loading buffer.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde at room temperature for 30 min and washed with PBS three times. Permeabilization was performed using 0.3% Triton X-100 in PBS for 30 min at room temperature, followed by blocking using 5% BSA (GC305010; Wuhan Servicebio Technology Co., Ltd.) solution supplemented with 0.3% Triton X-100 for 1 h at room temperature. Cells were then incubated with anti-RBM8A antibody (1:500; ab181038; Abcam) at 4°C overnight. After washing five times with PBS, the cells were incubated with FITC-labelled goat anti-rabbit secondary antibody (1:300; SA00003-2; Proteintech Group, Inc.) for 1 h at room temperature. After staining the nucleus with DAPI (1 μg/ml; C1006; Beyotime Institute of Biotechnology) at room temperature for 5 min, the cells were washed five times with PBS. Immunofluorescence signals were captured using a confocal laser scanning microscope (TCS SP8 DIVE; Leica Microsystems GmbH).
Fluorescence in situ hybridization (FISH) assay
AGS and HGC27 cells were seeded on sterile glass coverslips (801007; Wuxi NEST Biotechnology Co., Ltd.) and cultured for 48 h. The cell slides were washed three times with PBS and fixed with 4% paraformaldehyde at room temperature for 30 min. Subsequently, 2X saline-sodium citrate (SSC) buffer was used to wash the cells, and then cells were permeabilized with 2X SSC buffer containing 0.3% Triton X-100. Following permeabilization, cells were post-fixed with 4% paraformaldehyde in PBS for 5 min at room temperature to stabilize membrane integrity. The cells were pre-hybridized using pre-hybridization solution (G3047; Wuhan Servicebio Technology Co., Ltd.) containing 30% formamide at 50°C for 60 min. Cells were then hybridized with 1 μM BBC3 mRNA probes labelled with Cy5 (UCCAUCAGCCGUCCCUCUCCUGGCUUCUUG-Cy5) or 1 μM control probe (CAAGAAGCCAGGAGAGGGACGGCUGAUGGA-Cy5), which were synthesized by Sangon Biotech Co., Ltd., for 1 h at 50°C. Subsequently, 4X SSC buffer, 2X SSC buffer and 1X SSC buffer were used to wash the cells at 42°C for 5 min sequentially once each to remove excess probes. The cells were stained with DAPI for 5 min at room temperature. Immunofluorescence signals were captured using a confocal laser scanning microscope (TCS SP8 DIVE; Leica Microsystems GmbH).
Act D treatment assay (mRNA degradation assay)
AGS and HGC27 cells were transfected with siNC or siRBM8A using Lipofectamine 2000 according to the manufacturer's instructions as aforementioned. After 48 h of transfection, cells were treated with 5 μM Act D (HY-17559; MedChemExpress) dissolved in DMSO (D8371; Beijing Solarbio Science & Technology Co., Ltd.) or DMSO alone (same volume as Act D solution) as the control group for 0, 2, 4 or 6 h at 37°C. Total RNA was then extracted as aforementioned, and BBC3 mRNA expression was measured by qPCR as aforementioned.
Lentivirus packaging and cell transduction
Lentiviruses were packaged using a third-generation lentiviral system in 293T cells. A total of 15 μg of plasmid DNA was used for co-transfection per 10-cm dish, with the following ratio: 10 μg lentiviral vector (shRBM8A or shNC), 3 μg pGag/Pol (V017774; NovoPro), 1.5 μg pRev (V000387; NovoPro) and 0.5 μg pVsvg (V013596; NovoPro). Transfection was performed using Lipofectamine 2000 reagent (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol at 37°C for 6 h. Supernatants containing lentiviral particles were collected 48 h post-transfection and concentrated via 10% PEG6000 precipitation. AGS, HGC27 and MKN45 cells were transduced with lentiviruses [short hairpin RNA (shRNA/sh)RBM8A or shNC] at a multiplicity of infection of 10 in the presence of 8 μg/ml polybrene for 24 h. After transduction, cells were cultured in fresh medium for 24 h before selection. Stably transduced cells were selected with 2 μg/ml puromycin for 10 days, replacing the medium every 2-3 days. Following selection, cells were maintained in medium containing 1 μg/ml puromycin. Cells were allowed to recover for at least 7 days after the completion of selection before subsequent experiments to ensure stable knockdown. The target sequences of shRBM8A and shNC are listed in Table I.
Xenograft assay
MKN45 cells with stable knockdown of RBM8A (shRBM8A) and control cells (shNC) were constructed. According to the sample size calculation of G*Power 3.1.9.7 software (https://www.psychologie.hhu.de/), a total of 3 female 4-week-old BALB/c nude mice (14-16 g) were obtained from GemPharmatech Co., Ltd., via the Laboratory Animal Center of Xi'an Jiaotong University (Xi'an, China). Mice were housed in a specific pathogen-free (SPF) environment with ad libitum access to autoclaved food and water under controlled conditions: 23°C with 50% humidity and a 12-h light/dark cycle for a week. After acclimating to the SPF environment for a week, cells were resuspended in sterile PBS at a concentration of 5×106 cells per 100 μl and were injected subcutaneously into the bilateral axilla of nude mice (n=3/group): shNC cells were injected into the right axilla and shRBM8A cells were injected into the left axilla, with 5×106 cells per site. From 12 days after cell injection, the volume of subcutaneous tumors was measured using calipers every 2 days and calculated as 0.5 × length × width2. After 3 weeks of measurement, the nude mice were euthanized by intraperitoneal injection of sodium pentobarbital (150 mg/kg body weight), which is a commonly used and humane dose, to ensure a rapid and pain-free death. After injection, death was confirmed when there was a complete absence of respiratory movements for ≥5 min. Once the mice were confirmed to be euthanized, the subcutaneous tumors were removed and weighed. The investigator involved in cell injection, tumor volume measurement and data analysis was blinded to the group assignment. All animal care and procedures were conducted according to the guidelines of the National Institutes of Health and approved by Xi'an Jiaotong University Animal Care and Use Committee (approval no. 2024-1655; Xi'an, China).
IHC assay
Subcutaneous tumor tissues were fixed in 4% paraformaldehyde at room temperature for 24 h, followed by dehydration using a graded ethanol series (70, 80, 90, 95 and 100%) and embedding in paraffin blocks. Serial sections (5-μm-thick) were cut using a microtome (Leica RM2235; Leica Biosystems) and mounted on poly-L-lysine-coated slides. Slides were deparaffinized in xylene (2×10 min) and rehydrated through a descending ethanol series (100, 95, 80 and 70%) for 5 min each, followed by washing in distilled water. Antigen retrieval was performed by boiling sections in 10 mM sodium citrate buffer (pH 6.0) for 15 min using a pressure cooker. Sections were incubated in permeabilization buffer (0.3% Triton X-100 in PBS) for 15 min at room temperature. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 15 min at room temperature. Slides were then washed three times with PBS (pH 7.4) for 5 min each. Blocking was carried out using 5% BSA in PBS for 1 h at room temperature to minimize non-specific binding. Sections were incubated overnight at 4°C with rabbit polyclonal anti-RBM8A antibody (1:500 dilution; ab181038; Abcam) diluted in 1% BSA/PBS. After three washes with PBS, sections were incubated with HRP-conjugated goat anti-rabbit secondary antibody (1:500 dilution; ab6721; Abcam) for 1 h at room temperature. Signal detection was performed using a 3,3′-diaminobenzidine substrate kit (SK-4100; Vector Laboratories, Inc.) according to the manufacturer's instructions. Sections were counterstained with Mayer's hematoxylin for 30 sec at room temperature, dehydrated using ethanol gradients, cleared in xylene and mounted with coverslips using DPX mounting medium (MM1410; Maokang Biotechnology Co., Ltd.). Images of the cells were captured using an inverted fluorescence microscope (DMi8; Leica Microsystems GmbH) with bright-field imaging.
Statistical analysis
Statistical analysis of all experiments was performed using GraphPad Prism 8.0 (Dotmatics). All in vitro experiments were conducted in biological triplicates. Data are presented as the mean ± SD. Differences between groups were assessed using Student's t-test (a paired t-test was applied to evaluate the changes within the same cohort of subjects, whereas an unpaired t-test was used to compare the means between two distinct independent groups) or one-way ANOVA (Dunnett's multiple comparisons test was applied when comparing each experimental group against a single control group, and Tukey's Honestly Significant Difference test was used for pairwise comparisons among all experimental groups to assess differences between every possible group combination). Survival rates were evaluated using the Kaplan-Meier method and compared using the log-rank test. P<0.05 was considered to indicate a statistically significant difference.
Results
RBM8A expression is upregulated in GC tissues
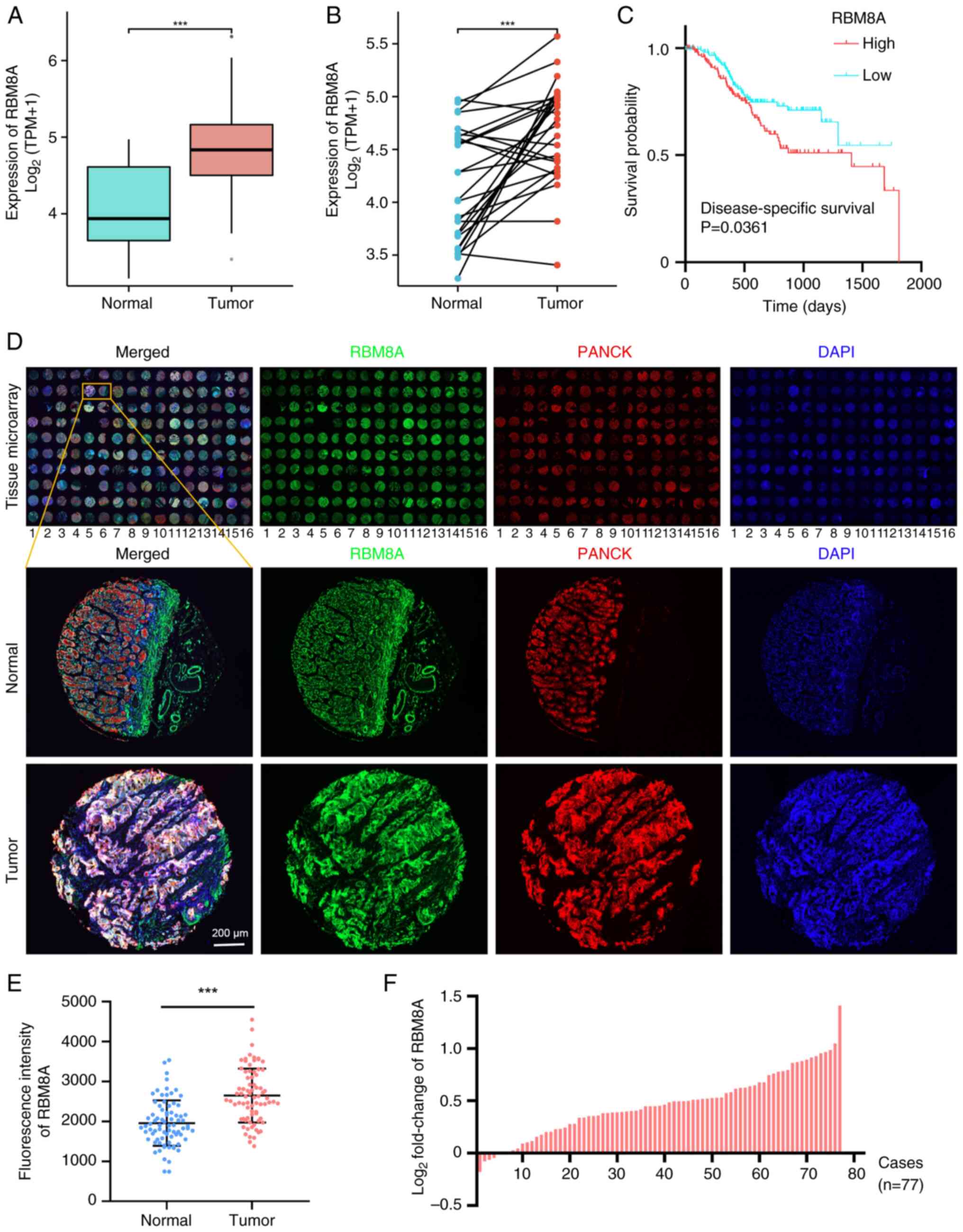
Transcriptome sequencing analysis of GC in TCGA demonstrated that the expression levels of RBM8A in GC tissues were significantly higher than those in normal tissues (Fig. 1A and B). Furthermore, high RBM8A expression was associated with a poor prognosis (Fig. 1C). This is consistent with the data reported by Lv and Cheng (23). Additionally, mIHC was used to detect the expression levels of RBM8A in epithelial cells (PANCK+ cells) of a GC tissue microarray (n=80) (Fig. 1D). A total of 3 pairs of samples were lost due to tissue sample debridement during the operation. The analysis results showed that RBM8A expression was specifically and significantly on average 1.4-fold higher (P<0.05) in tumor tissues, with a maximum log2 fold change of 1.4 (2.6-fold increase), compared with in adjacent normal tissues (Fig. 1E and F). These results suggested that RBM8A serves a pro-cancer role in GC.
Knockdown of RBM8A inhibits GC cell proliferation and promotes apoptosis in vitro
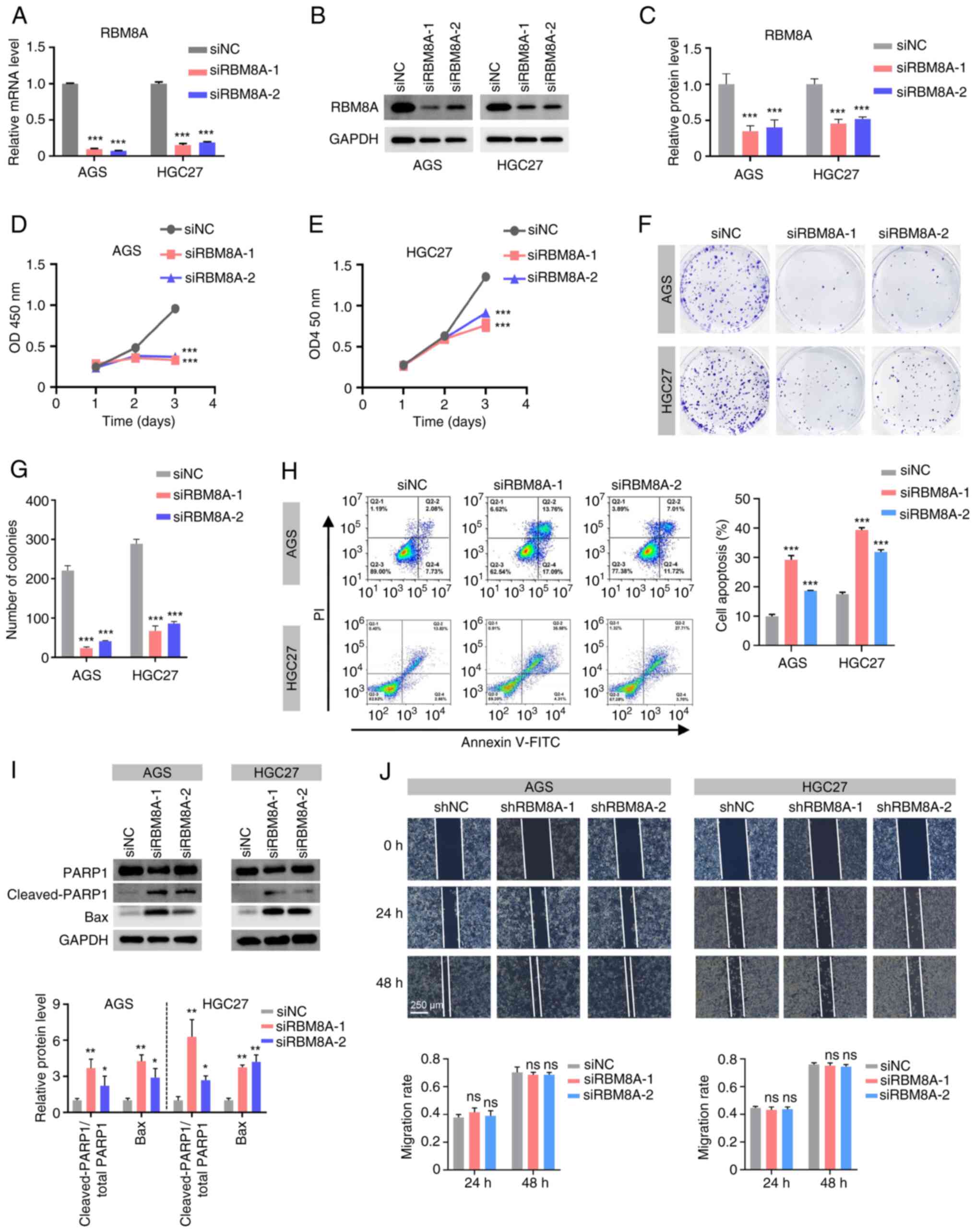
To explore the cellular biological functions through which RBM8A promotes GC progression, its biological roles were investigated in AGS and HGC27 cells. Firstly, siRNAs for RBM8A were designed. qPCR results indicated that in AGS cells, siRBM8A-1 and siRBM8A-2 decreased the mRNA levels of RBM8A by 90.4 and 92.5%, respectively, while in HGC27 cells, they reduced the mRNA levels by 84.9 and 81.5%, respectively (Fig. 2A). The results of western blotting demonstrated that siRBM8A-1 and siRBM8A-2 reduced the RBM8A protein levels in AGS cells by 65.1 and 59.8%, respectively, and in HGC27 cells by 54.5 and 48.5%, respectively (Fig. 2B and C). These results demonstrated that the designed siRBM8As were able to effectively knock down RBM8A at both the mRNA and protein level. Subsequently, functional assays were performed to assess the impact of RBM8A knockdown on GC cell behavior. The CCK8 assay revealed a significant reduction in GC cell proliferation at 72 h following RBM8A knockdown (Fig. 2D and E), while the colony formation assay further demonstrated that knockdown of RBM8A markedly suppressed the colony formation of GC cells (Fig. 2F and G). Furthermore, the results of flow cytometry showed that transfection with siRBM8A-1 or siRBM8A-2 led to a 2.9- or 1.9-fold increase in the apoptosis rate of AGS cells, and a 2.3- or 1.8-fold increase in the apoptosis rate of HGC27 cells, respectively (Fig. 2H). Western blotting showed that transfection of these two siRBM8As into AGS cells significantly upregulated the ratio of cleaved PARP1 to total PARP1 by 3.7- and 2.2-fold, respectively, and the levels of Bax by 4.3- and 2.9-fold, respectively (Fig. 2I). Similarly, in HGC27 cells, the ratio of the cleaved PARP1 to total PARP1 was upregulated by 6.3- and 2.7-fold, respectively, and Bax expression was upregulated by 3.7- and 4.2-fold, respectively (Fig. 2I). To examine cell migration, AGS and HGC27 cell lines with stable knockdown of RBM8A were established. qPCR showed that shRBM8A-1 and shRBM8A-2 decreased the mRNA levels of RBM8A in AGS cells by 67.0 and 50.5%, respectively, while in HGC27 cells, they reduced the mRNA levels by 63.3 and 49.2%, respectively (Fig. S1A and B). By contrast, the cell scratch assay revealed that knockdown of RBM8A had no marked effect on cell migration (Fig. 2J). Taken together, these findings suggested that RBM8A serves a crucial role in promoting GC progression primarily by inhibiting cell apoptosis, rather than influencing cell migration.
Knockdown of RBM8A inhibits GC tumor progression in a xenograft model
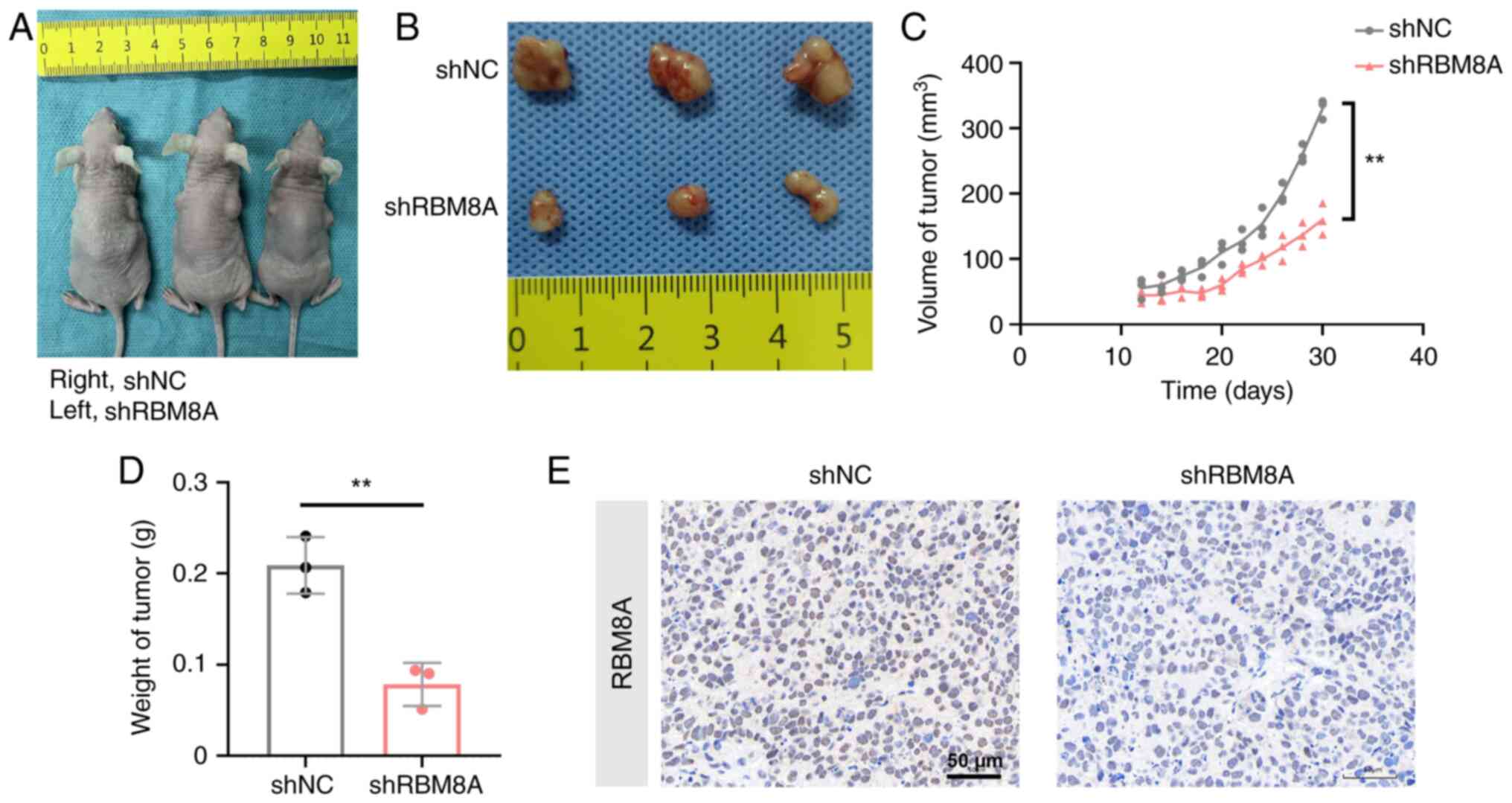
To examine the roles of RBM8A in tumorigenesis in vivo, MKN45 cell lines with stable knockdown of RBM8A and control cell lines were constructed, and Fig. S1C shows that shRBM8A effectively decreased RBM8A expression to 42.3% compared with shNC. MKN45 cells stably expressing shRBM8A or shNC were subcutaneously injected into nude mice (Fig. 3A and B). The maximum tumor volume reached 341.4 mm3, and the maximum tumor diameter was 9.96 mm. This tumor size complies with the animal ethics guidelines and welfare standards approved by our institutional review board. Both the size (Fig. 3C) and weight (Fig. 3D) of the tumors in the shRBM8A group were reduced by 51.5 and 62.4% compared with those in the shNC group, respectively, when measured at 32 days after cell inoculation. Immunohistochemistry also showed downregulation of RBM8A expression in the shRBM8A group (Fig. 3E). These results suggested that RBM8A promoted GC cell proliferation in vivo.
mRNA-seq analysis of DEGs regulated by RBM8A
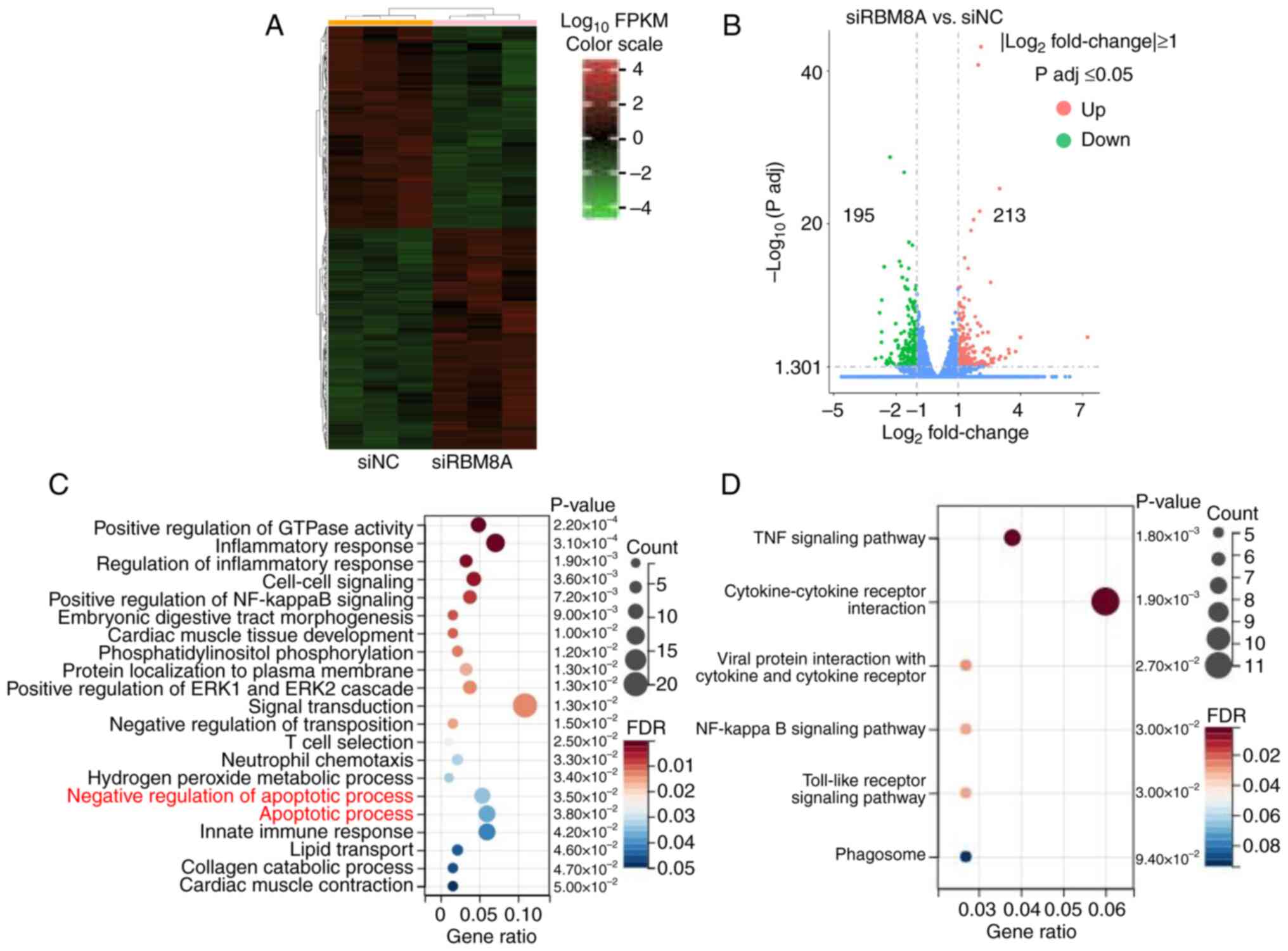
To further determine the molecular mechanism by which RBM8A regulates GC cell apoptosis, the transcriptome of AGS cells following RBM8A knockdown was examined using mRNA-seq. Bioinformatics analysis identified a total of 408 DEGs following RBM8A knockdown, with 213 upregulated genes and 195 downregulated genes (Fig. 4A and B). Further functional enrichment analysis provided valuable insights into the biological processes influenced by RBM8A. GO analysis indicated that these DEGs were enriched in terms such as 'positive regulation of GTPase activity', 'inflammatory response', 'cell-cell signaling' and 'apoptotic process' (Fig. 4C). KEGG pathway analysis revealed that these DEGs were enriched in 'TNF signaling pathway', 'cytokine-cytokine receptor interaction', 'NF-kappa B signaling pathway' and 'Toll-like receptor signaling pathway' (Fig. 4D), all of which serve key roles in apoptosis regulation. Taken together, these findings further support the role of RBM8A in apoptosis regulation. However, to fully elucidate its molecular mechanism, additional studies are required to identify the direct target genes regulated by RBM8A.
RIP-seq analysis of RBM8A-interacting mRNAs
As an RNA-binding protein, RBM8A is expected to interact with specific mRNAs to exert its regulatory functions. To further investigate its direct target genes, RIP-seq analysis was used to identify mRNAs bound by RBM8A. This analysis identified a total of 2,079 potential target genes directly interacting with RBM8A (Fig. 5A). Notably, these potential target genes are distributed on all chromosomes of the genome (Fig. 5B). Further analysis of the binding regions showed that RBM8A binds to multiple genomic elements, including the promoter, UTRs, intron, and other exon or intron regions of its target genes (Fig. 5C). To gain deeper insights into the biological significance of these interactions, GO enrichment analysis was performed. The results demonstrated that RBM8A-bound genes were significantly associated with various biological processes, including 'membrane-enclosed lumen', 'nucleoplasm', 'RNA binding' and 'apoptotic process' (Fig. 5D). The enrichment of apoptosis-related genes aligns well with the present in vitro findings, further reinforcing the role of RBM8A in apoptosis regulation. To identify key target genes, combined analysis of mRNA-seq and RIP-seq data was conducted. Through this integrative approach, eight genes were identified as potential targets of RBM8A, including S100A16, BBC3, AC073410.1, GAS5, CCL4L2, STX16-NPEPL1, POLR2J3 and HIGD1A (Fig. 5E). Among these, BBC3, a well-known pro-apoptotic gene (24,25), stood out as a particularly relevant candidate. qPCR validation demonstrated that the mRNA levels of BBC3 were significantly upregulated following RBM8A knockdown in both AGS and HGC27 cells (Fig. 5F). Taken together, these results suggested that RBM8A may regulate the apoptosis of GC cells by interacting with BBC3 mRNA and affecting its stability. However, whether BBC3 mediates RBM8A-regulated apoptosis and the direct interaction between RBM8A and BBC3 mRNA still require further experimental verification.
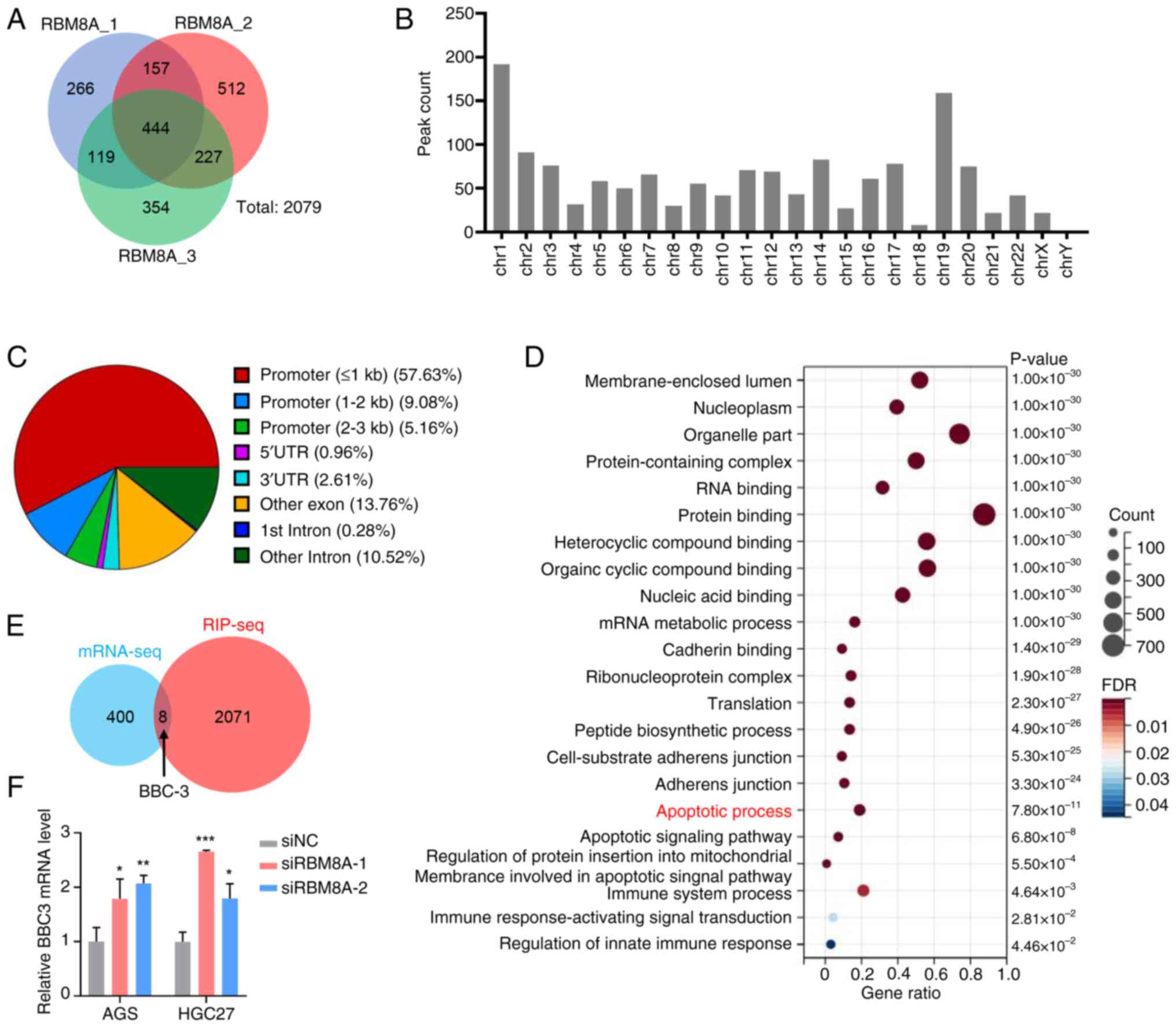
BBC3 mediates the effect of RBM8A on the regulation of apoptosis in GC cells
To further investigate whether BBC3 serves a mediating role in RBM8A-regulated apoptosis, rescue experiments were conducted in AGS and HGC27 cells. First, BBC3 siRNA was designed and its knockdown efficiency was detected by qPCR and western blotting (Fig. 6A and B). Next, siRBM8A and siBBC3 were co-transfected into GC cells, and the BBC3 mRNA levels were subsequently detected. Compared with the siNC control group, transfection with siRBM8A significantly upregulated BBC3 mRNA levels. By contrast, co-transfection with siRBM8A and siBBC3 significantly downregulated BBC3 mRNA levels compared with the siRBM8A single-transfection group (Fig. 6C). Functional assays further demonstrated the regulatory relationship between these two genes. The CCK8 assay results indicated that knockdown of BBC3 partially reversed the inhibitory effect of RBM8A depletion on cell proliferation (Fig. 6D). Consistently, flow cytometry revealed that BBC3 knockdown diminished the RBM8A-knockdown-induced apoptosis upregulation from 5.2- to 1.3-fold in AGS cells and from 2.5-fold to 1.2-fold in HGC27 cells (Fig. 6E). These results indicated that knockdown of BBC3 could partially reverse the pro-apoptotic effect of RBM8A depletion in GC cells. The aforementioned results confirmed that BBC3 mediated the inhibition of apoptosis by RBM8A in GC cells. RBM8A exerts its anti-apoptotic effects in GC cells by modulating BBC3 expression, highlighting a potential molecular mechanism underlying GC progression.
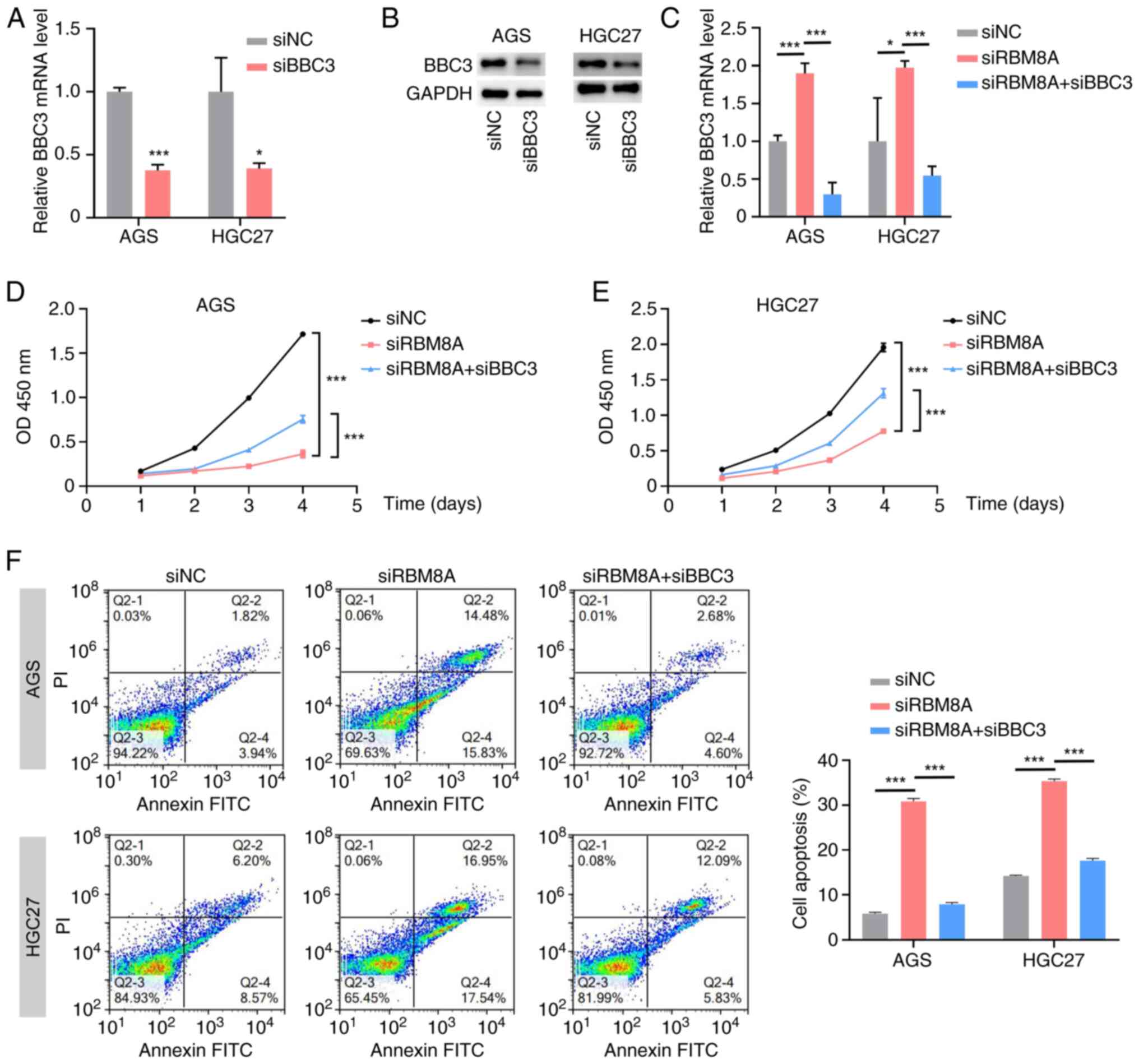
RBM8A directly binds to BBC3 mRNA and regulates its stability
To verify that RBM8A directly binds to BBC3 mRNA and modulates its stability, RIP-qPCR and agarose gel electrophoresis were first performed. These experiments demonstrated that the RBM8A antibody successfully enriched BBC3 mRNA (Fig. 7A-C), providing initial evidence of their direct interaction. In addition, a probe for BBC3 mRNA was designed. The RNA pulldown assay results demonstrated that the probe could enrich both BBC3 mRNA and RBM8A protein (Fig. 7D and E). Furthermore, the results of FISH-immunofluorescence (IF) revealed that RBM8A and BBC3 mRNA were co-localized in AGS and HGC27 cells (Fig. 7F). These results demonstrated the direct binding of RBM8A to BBC3 mRNA. Having established their binding relationship, the present study subsequently explored the functional effect of RBM8A on BBC3 mRNA stability. Actinomycin D was used to treat GC cells in a time-dependent manner to inhibit transcription. Simultaneously, RBM8A was knocked down in GC cells to explore the degradation rate of BBC3 after knockdown of RBM8A. qPCR demonstrated that following transcription inhibition, the BBC3 mRNA level was significantly downregulated (Fig. 7G and H). Furthermore, compared with that in the control group transfected with siNC, the downregulation rate of BBC3 mRNA in cells with knockdown of RBM8A was slower, indicating that knockdown of RBM8A inhibited the degradation of BBC3 (Fig. 7G and H). Collectively, these findings suggested that RBM8A promoted the degradation of mRNA of the pro-apoptotic gene BBC3 by directly binding to it, thereby inhibiting the apoptosis of GC cells. This mechanistic insight highlights the crucial role of RBM8A in GC progression.
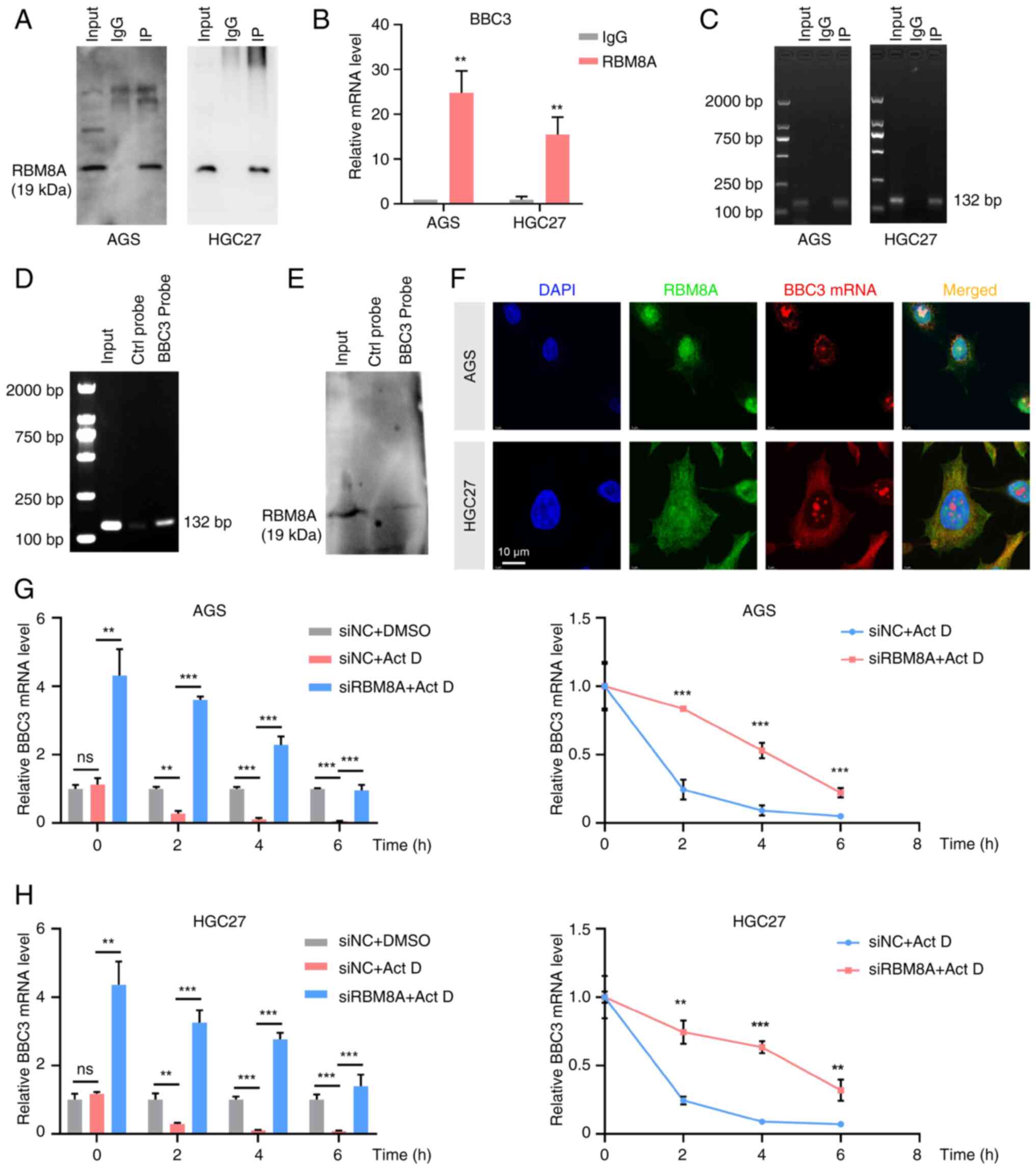
RBM8A interacts with UPF3B to regulate BBC3 mRNA stability and apoptosis
To further elucidate the potential molecular mechanism by which RBM8A regulates the stability of BBC3 mRNA, CoIP and protein MS were conducted to identify the proteins that interact with RBM8A. A total of 416 proteins that might interact with RBM8A were identified in the protein MS experiment. GO analysis of the genes corresponding to these proteins showed enrichment in biological processes such as 'cytoplasmic translation', 'RNA splicing, via spliceosome' and 'regulation of mRNA stability' (Fig. 8A). Notably, the term 'negative regulation of apoptotic process' was also enriched (Fig. 8A), aligning with our aforementioned in vitro findings indicating that RBM8A serves a role in apoptosis regulation.
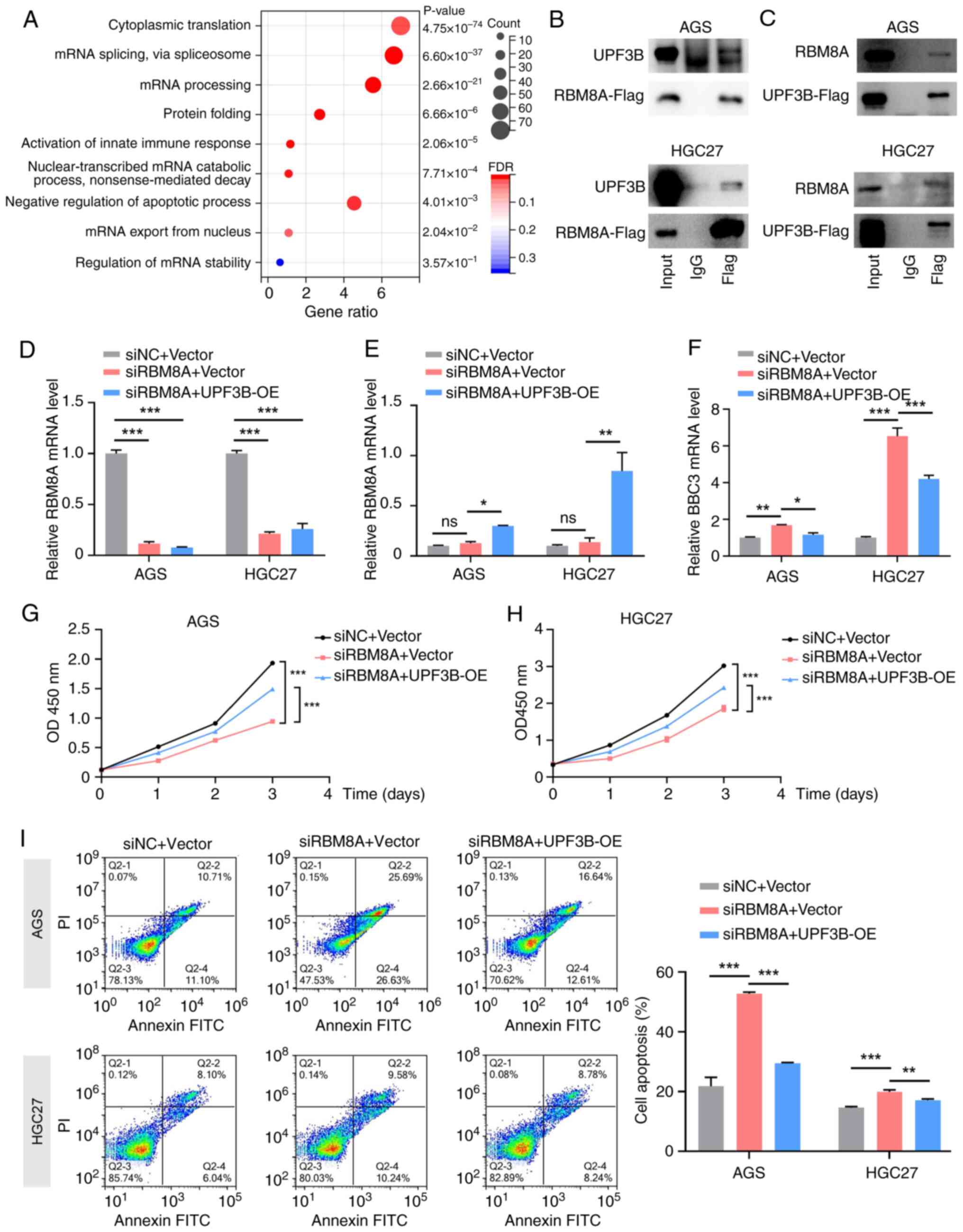
In the term 'regulation of mRNA stability', UPF3B was identified as a potential RBM8A-interacting protein. Subsequently, their interaction was further verified by CoIP-western blotting. The results demonstrated that following overexpression of RBM8A tagged with 3X Flag (RBM8A-3X Flag), the Flag antibody could enrich RBM8A and UPF3B in AGS and HGC27 GC cells (Fig. 8B). Conversely, following overexpression of UPF3B tagged with 3X Flag (UPF3B-3X Flag) in AGS and HGC27 GC cells, the Flag antibody could also enrich UPF3B and RBM8A (Fig. 8C). These results indicated an interaction between RBM8A and UPF3B. Subsequently, it was examined whether UPF3B mediates the regulation of BBC3 mRNA. Firstly, the transfection efficiency of the UPF3B overexpression vector in AGS and HGC27 cells was detected. qPCR demonstrated that the mRNA levels of UPF3B were significantly upregulated after transfection with the UPF3B overexpression vector (Fig. S1D). Subsequently, qPCR analysis revealed that overexpression of UPF3B counteracted the upregulation of BBC3 mRNA induced by RBM8A knockdown in AGS and HGC27 GC cells (Fig. 8D-F). These findings suggested that UPF3B serves a crucial role in modulating BBC3 mRNA levels in response to RBM8A depletion.
The present study further investigated whether UPF3B mediated the biological functions of RBM8A. CCK8 assay results indicated that overexpression of UPF3B partially reversed the proliferation inhibition caused by RBM8A knockdown (Fig. 8G and H). Similarly, flow cytometry revealed that overexpression of UPF3B could partially attenuate the pro-apoptotic effects of RBM8A knockdown (Fig. 8I). Collectively, these results confirmed that UPF3B mediated the inhibition of apoptosis by RBM8A in GC cells. The findings further highlighted that RBM8A promoted GC progression by interacting with UPF3B to regulate BBC3 mRNA stability.
Discussion
GC is one of the most common malignant tumors in the world, and the incidence and mortality rates of GC are among the highest in China (26). Early symptoms are not obvious and there is a lack of effective early diagnostic models, resulting in most patients with GC being in the advanced stage of cancer when diagnosed, which leads to a low 5-year survival rate and poor prognosis of patients (2). Therefore, there is an urgent need for in-depth research on GC-targeted therapeutic molecules to enhance patient survival.
Current oncology research primarily focuses on tumor immunity, cellular metabolism, epigenetic modification, non-coding RNA and mRNA metabolic regulation. Dysregulation of mRNA metabolism is a fundamental trait of tumorigenesis (27). A growing number of large-scale genomic investigations have revealed that the disruption of post-transcriptional gene expression regulation can fuel cancer progression (28-30). These regulatory alterations span across every stage of RNA processing, including mRNA splicing, transportation, editing and degradation (27). The identification of these phenomena has shed light on the adverse effects of aberrant RNA processing in cancer on numerous novel treatment strategies and has opened up novel avenues for cancer treatment (27). A number of RNA-binding proteins have a major impact on GC tumorigenesis by regulating a number of mRNAs at the post-transcriptional level. For example, pumilio RNA binding family member 1 promotes GC progression by binding to the DEP domain containing MTOR interacting protein mRNA pumilio response element to maintain its stability, which in turn inhibits activation of PI3K/AKT signaling and glycolysis by mTORC1 (28). Epithelial splicing regulatory protein 1 promotes exon 11-selective splicing of calsyntenin 1 (CLSTN1) precursor mRNA. The short spliced CLSTN1 stabilizes the E-calmodulin/β-catenin binding structure and promotes ubiquitination and degradation of β-catenin, thereby inhibiting migration and invasion of GC cells (29). Karyopherin subunit α2 promotes GC cell proliferation, migration and invasion by regulating the alternative 3′-splice site selection of WD repeat domain 62 (30). The present study revealed that RBM8A expression was significantly upregulated in GC tissues, and knockdown of RBM8A in GC cells significantly inhibited GC cell proliferation and promoted apoptosis. These findings demonstrated its pro-cancer role in GC, and future studies are needed to explore the role of RBM8A in different cancer contexts.
To further explore the molecular mechanism of RBM8A-promoted GC progression, RIP-seq was conducted to identify RNAs that bind to the RBM8A protein. Through combined analysis with mRNA-seq data, BBC3 was identified. BBC3, also known as p53 upregulated modulator of apoptosis, encodes a member of the BCL-2 protein family. As a BH3-only pro-apoptotic subclass member, BBC3 can directly bind to anti-apoptotic BCL-2 family members, mediating cell apoptosis (31). This leads to the translocation of Bax and/or BCL2 antagonist/killer proteins, their activation and the formation of pore-like structures on the mitochondrial membrane, causing mitochondrial dysfunction, caspase activation and apoptosis (32-34). BBC3 inhibits hepatocellular carcinoma (HCC) cell apoptosis, and thus, promotes HCC progression, an effect negatively regulated by microRNA-222 (35). In GC, interferon regulatory factor 1 activates BBC3 transcription and induces cell apoptosis via the intrinsic pathway independent of the extrinsic pathway (20). The present results demonstrated that knockdown of RBM8A significantly enhanced BBC3 expression, and the pro-apoptotic protein Bax was also significantly upregulated. Based on previous findings (32,33,35) and our experimental results, RBM8A may inhibit GC cell apoptosis via post-transcriptional regulation of BBC3 expression, thereby relieving BBC3-mediated activation of Bax. In terms of the molecular mechanism by which RBM8A regulates BBC3, the specificity of the binding between RBM8A and BBC3 mRNA is an important aspect that requires further investigation. In the current study, the interaction between RBM8A and BBC3 mRNA was mainly demonstrated through RIP-qPCR and FISH-IF. However, these experiments still have limitations. First, the specific binding between RBM8A and BBC3 mRNA has not been directly validated by in vitro competition experiments to test binding specificity. Second, based on our screening threshold, only eight target genes were screened out in the combined analysis of RIP-seq and mRNA-seq. This may be related to the screening threshold or the fact that RBM8A binding to these target gene mRNAs may not directly affect their stability but rather regulates processes such as translation and nuclear export. Further studies are required to verify this by analyzing downstream target gene protein levels.
A previous study has reported that RBM8A and UPF3B serve important roles in gene regulation as essential components of the EJC complex (36). UPF3B functions as an adaptor protein, serving concomitantly as an nMD amplifier and an nMD branch-specific factor. In the preponderantly acknowledged nMD model, UPF3B, in its capacity as an adaptor, engenders macromolecular complexes that instigate RNA decay (37,38). Upon interaction between the EJC and the nMD factor UPF3B, the process of RNA decay is initiated (39,40). Initially, nMD was considered to be a quality control apparatus capable of promptly degrading abnormal mRNAs harboring premature termination codons. However, evidence has revealed that nMD also targets numerous normal mRNAs for degradation (41), functioning similarly to transcription factors that modulate RNA synthesis rates of specific gene subsets (41,42). nMD governs the stability of particular subsets of normal mRNAs (42). While the full physiological significance of this process remains indeterminate, an accumulating corpus of evidence suggests that nMD represents a highly regulated pathway that selectively degrades subsets of mRNAs, thereby influencing specific biological events (41,43). UPF3B is upregulated in HCC samples and is associated with poor prognosis in patients. UPF3B binds to protein phosphatase 2 regulatory subunit Bγ, promotes its mRNA degradation and activates the PI3K/AKT/mTOR pathway, thereby promoting HCC cell proliferation (44). In addition, a truncated splice variant of UPF3B, UPF3B-S, exists in HCC cells, and this promotes HCC migration and invasion by targeting the 3′UTR of cadherin 1 mRNA to enhance its degradation, leading to activation of epithelial-mesenchymal transition (45). Given these findings, the present study aimed to investigate whether UPF3B and RBM8A possess similar biological functions in GC. Using CoIP, it was demonstrated that RBM8A and UPF3B also formed a complex in GC. Additionally, UPF3B overexpression reversed the RBM8A knockdown-mediated upregulation of BBC3 mRNA in GC cells. These results demonstrated that RBM8A promoted GC progression by interacting with UPF3B to regulate BBC3 mRNA stability. Compared with a previous study by Lv and Cheng (23), which primarily identified the role of RBM8A in GC prognosis, the present study identified BBC3 as a novel target of RBM8A and elucidated the role of UPF3B in the RBM8A-mediated regulation of BBC3. These findings expand the understanding of the molecular mechanisms underlying RBM8A-promoted GC progression via inhibition of cell apoptosis.
The present study has several limitations. First, the present study mainly relied on in vitro cell line and xenograft models, which may not fully recapitulate the complexity of human GC. Future research could incorporate patient-derived organoids to better mimic the in vivo microenvironment. Second, the present findings lack clinical validation. Large-scale patient cohort studies are needed to confirm the role of RBM8A and BBC3 in patients with GC. Additionally, exploring serum RBM8A as a biomarker could be a potential direction. Third, although RBM8A was knocked down using shRNA/siRNA, potential off-target effects cannot be completely excluded. Further studies are required to verify the specificity of RBM8A knockdown. In terms of therapeutic implications, targeting RBM8A could be a potential strategy for the treatment of GC. Small molecule inhibitors of RBM8A or antisense oligonucleotides could be explored to inhibit its function in preclinical studies. However, several challenges remain. For instance, it is still unclear whether small molecule inhibitors of RBM8A can be designed to effectively suppress the function of RBM8A. Additionally, there is a need to figure out how to precisely deliver these small molecule inhibitors or antisense oligonucleotides to the tumor site rather than other tissues. In addition, since RBM8A is ubiquitously expressed (15), the impact of inhibiting its function on normal tissue cells requires further investigation to explore the potential toxicity of drugs targeting RBM8A. Further research is needed to optimize the drug-delivery methods and evaluate the safety of these approaches. Furthermore, it has been reported that RBM8A serves an important role in breast cancer cisplatin resistance (19). Further studies are warranted to determine whether RBM8A contributes to resistance to drugs other than cisplatin in GC. A comprehensive analysis of the interaction between RBM8A expression in tumors or body fluids and the efficacy of various cancer therapies holds great potential for advancing precision medicine and optimizing therapeutic strategies. The present GO analysis of both mRNA-seq and RIP-seq data showed that RBM8A-regulated differential genes or mRNAs bound to RBM8A could be enriched in terms such as 'inflammatory response', 'innate immune response' and 'cell-cell signaling', suggesting that RBM8A may also modulate the tumor microenvironment of GC by regulating cytokine secretion and may be involved in immune escape in GC; however, these hypotheses need to be verified by further experiments.
In summary, the present study revealed that RBM8A promoted the mRNA degradation of the pro-apoptotic gene BBC3 by interacting with UPF3B, thereby inhibiting cell apoptosis and promoting GC progression. The identification of BBC3 as a novel target and the role of UPF3B in this process are important contributions to the understanding of RBM8A-mediated GC progression mechanisms. These findings reveal approaches with translational potential for GC treatment, including inhibition of RBM8A expression or inhibition of RBM8A function, including binding to BBC3 mRNA and interaction with UPF3B, via different means such as antisense oligonucleotides and small molecule inhibitors; however, further validation through clinical trials and in-depth mechanistic studies is required.
Supplementary Data
Availability of data and materials
The RIP-seq data generated in the present study may be found in the Gene Expression Omnibus database under accession number GSE292233 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE292233. The mRNA-seq data generated in the present study may be found in the Gene Expression Omnibus database under accession number GSE292234 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE292234. The CoIP-MS data generated in the present study may be found in the iProX repository under accession number PXD063845 or at the following URL: https://www.iprox.cn/page/project.html?id=IPX0011930000 (46,47). The other data generated in the present study may be requested from the corresponding author.
Authors' contributions
JW and CH conceived and designed the study. HP, FL, XJ and LC performed all in vitro experiments. CX provided support with experimental techniques. LZ performed the bioinformatics analysis. LZ and JZ performed the in vivo experiments. HP and LZ were major contributors in writing the manuscript. FL and JZ confirm the authenticity of all the raw data. JW, HP and LZ provided financial support. All authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
The animal experiments in the present study were approved by the Xi'an Jiaotong University Animal Care and Use Committee (approval no. 2024-1165; Xi'an, China). The use of human tissue microarrays was reviewed and approved by the Medical Ethics Committee of Shaanxi Provincial People's Hospital (approval no. 2023-R176; Xi'an, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
The authors would like to thank the laboratory of the Biomedical Experimental Center of the Department of Medicine (Xi'an Jiaotong University, Xi'an, China) for providing the experimental platform for this study.
Funding
The present study was supported by the Technology Incubation Fund and Talent Program Projects of Shaanxi Provincial People's Hospital (grant no. 2021LJ-02), the Project of the Health Research and Innovation Capacity Enhancement Plan (grant no. 2024PT-01), Science and Technology Innovation Leading Talents of the 'Special Support Plan for High-level Talents' in Shaanxi Province (grant nos. 2022JY-44 and 2022YJY-30), the Shaanxi Postdoctoral Research Program (grant no. 2023BSHGZZH QYXMZZ34), The Xi'an Science and Technology Program (grant no. 24YXYJ0168), and the Shaanxi Provincial Natural Science Foundation Youth Program (grant nos. 2024JC-YBQN-0803 and 2025JC-YBQN-1116).
References
|
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 74:229–263. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Guan WL, He Y and Xu RH: Gastric cancer treatment: Recent progress and future perspectives. J Hematol Oncol. 16:572023. View Article : Google Scholar : PubMed/NCBI | |
|
Joshi SS and Badgwell BD: Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 71:264–279. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Chuang TW, Lee KM and Tarn WY: Function and pathological implications of exon junction complex factor Y14. Biomolecules. 5:343–355. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Bai R, Wan R, Wang L, Xu K, Zhang Q, Lei J and Shi Y: Structure of the activated human minor spliceosome. Science. 371:eabg08792021. View Article : Google Scholar : PubMed/NCBI | |
|
Zhan X, Yan C, Zhang X, Lei J and Shi Y: Structure of a human catalytic step I spliceosome. Science. 359:537–545. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Noble CG and Song H: mlN51 stimulates the RNA-helicase activity of eIF4AIII. PLoS One. 2:e3032007. View Article : Google Scholar : PubMed/NCBI | |
|
Hosoda N, Kim YK, Lejeune F and Maquat LE: CBP80 promotes interaction of Upf1 with Upf2 during nonsense-mediated mRNA decay in mammalian cells. Nat Struct Mol Biol. 12:893–901. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Isken O and Maquat LE: The multiple lives of nMD factors: Balancing roles in gene and genome regulation. Nat Rev Genet. 9:699–712. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Woeller CF, Gaspari M, Isken O and Maquat LE: nMD resulting from encephalomyocarditis virus IRES-directed translation initiation seems to be restricted to CBP80/20-bound mRNA. EMBO Rep. 9:446–451. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G and Ohno S: Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 20:355–367. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
López-Perrote A, Castaño R, Melero R, Zamarro T, Kurosawa H, Ohnishi T, Uchiyama A, Aoyagi K, Buchwald G, Kataoka N, et al: Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res. 44:1909–1923. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Behm-Ansmant I and Izaurralde E: Quality control of gene expression: A stepwise assembly pathway for the surveillance complex that triggers nonsense-mediated mRNA decay. Genes Dev. 20:391–398. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Kervestin S and Jacobson A: nMD: A multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 13:700–712. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Mei N, Chen H, Zhao N, Yi Y and Li C: A comprehensive pan-cancer analysis of RBM8A based on data mining. J Oncol. 2021:99833542021. View Article : Google Scholar : PubMed/NCBI | |
|
Lin Y, Wei L, Hu B, Zhang J, Wei J, Qian Z and Zou D: RBM8A promotes glioblastoma growth and invasion through the notch/STAT3 pathway. Front Oncol. 11:7369412021. View Article : Google Scholar : PubMed/NCBI | |
|
Li F, Wang X, Zhang J, Zhang J, Jing X, Jiang Q, Zhou J, Cao L, Peng H, Tong D and Huang C: RBM8A, a new target of TEAD4, promotes breast cancer progression by regulating IGF1R and IRS-2. J Transl Med. 22:8232024. View Article : Google Scholar : PubMed/NCBI | |
|
Liang R, Lin Y, Ye JZ, Yan XX, Liu ZH, Li YQ, Luo XL and Ye HH: High expression of RBM8A predicts poor patient prognosis and promotes tumor progression in hepatocellular carcinoma. Oncol Rep. 37:2167–2176. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Song T and Zhang H: RBM8A depletion decreases the cisplatin resistance and represses the proliferation and metastasis of breast cancer cells via AKT/mTOR pathway. Breast J. 2022:45767892022. View Article : Google Scholar : PubMed/NCBI | |
|
Gao J, Senthil M, Ren B, Yan J, Xing Q, Yu J, Zhang L and Yim JH: IRF-1 transcriptionally upregulates PUMA, which mediates the mitochondrial apoptotic pathway in IRF-1-induced apoptosis in cancer cells. Cell Death Differ. 17:699–709. 2010. View Article : Google Scholar | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar | |
|
Peng H, Zhang W, Dong H, Yuan J, Li Y, Li F, Yu D, Guan Y and Zhang F: CircFAT1 promotes lung adenocarcinoma progression by sequestering miR-7 from repressing IRS2-ERK-mediated CCND1 expression. Int J Biol Sci. 18:3944–3960. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Lv X and Cheng H: Prognostic value of increased expression of RBM8A in gastric cancer. Braz J Med Biol Res. 53:e92902020. View Article : Google Scholar : PubMed/NCBI | |
|
Yu J and Zhang L: No PUMA, no death: Implications for p53-dependent apoptosis. Cancer Cell. 4:248–249. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Li M: The role of P53 up-regulated modulator of apoptosis (PUMA) in ovarian development, cardiovascular and neurodegenerative diseases. Apoptosis. 26:235–247. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Qiu L, Yao L, Hu P and He T: Analysis of the detection rate and clinical characteristics of early gastric cancer by painless gastroscopy and ordinary gastroscopy. Medicine (Baltimore). 103:e381202024. View Article : Google Scholar : PubMed/NCBI | |
|
Obeng EA, Stewart C and Abdel-Wahab O: Altered RNA processing in cancer pathogenesis and therapy. Cancer Discov. 9:1493–1510. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Yin S, Liu H, Zhou Z, Xu X, Wang P, Chen W, Deng G, Wang H, Yu H, Gu L, et al: PUM1 promotes tumor progression by activating DEPTOR-meditated glycolysis in gastric cancer. Adv Sci (Weinh). 10:e23011902023. View Article : Google Scholar : PubMed/NCBI | |
|
Li C, Yin Y, Tao R, Lin Y, Wang T, Shen Q, Li R, Tao K and Liu W: ESRP1-driven alternative splicing of CLSTN1 inhibits the metastasis of gastric cancer. Cell Death Discov. 9:4642023. View Article : Google Scholar : PubMed/NCBI | |
|
Chen X, Wei H, Yue A, Zhang H, Zheng Y, Sun W, Zhou Y and Wang Y: KPNA2 promotes the progression of gastric cancer by regulating the alternative splicing of related genes. Sci Rep. 14:171402024. View Article : Google Scholar : PubMed/NCBI | |
|
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman pM, Day CL, Adams JM and Huang DC: Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 17:393–403. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR and Newmeyer DD: BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 17:525–535. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S and Korsmeyer SJ: Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2:183–192. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Mérino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD and Bouillet P: The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 186:355–362. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Z, Sun J, Liu B, Zhao M, Xing E and Dang C: miRNA-222 promotes liver cancer cell proliferation, migration and invasion and inhibits apoptosis by targeting BBC3. Int J Mol Med. 42:141–148. 2018.PubMed/NCBI | |
|
Buchwald G, Ebert J, Basquin C, Sauliere J, Jayachandran U, Bono F, Le Hir H and Conti E: Insights into the recruitment of the nMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc Natl Acad Sci USA. 107:10050–10055. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Celik A, Kervestin S and Jacobson A: nMD: At the crossroads between translation termination and ribosome recycling. Biochimie. 114:2–9. 2015. View Article : Google Scholar : | |
|
Chan WK, Huang L, Gudikote JP, Chang YF, Imam JS, MacLean JA II and Wilkinson MF: An alternative branch of the nonsense-mediated decay pathway. EMBO J. 26:1820–1830. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Chamieh H, Ballut L, Bonneau F and Le Hir H: nMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol. 15:85–93. 2008. View Article : Google Scholar | |
|
Jones SH and Wilkinson M: RNA decay, evolution, and the testis. RNA Biol. 14:146–155. 2017. View Article : Google Scholar : | |
|
Karam R, Wengrod J, Gardner LB and Wilkinson MF: Regulation of nonsense-mediated mRNA decay: Implications for physiology and disease. Biochim Biophys Acta. 1829:624–633. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Lykke-Andersen S and Jensen TH: Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 16:665–677. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Goetz AE and Wilkinson M: Stress and the nonsense-mediated RNA decay pathway. Cell Mol Life Sci. 74:3509–3531. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Hou B, Shu M, Liu C, Du Y, Xu C, Jiang H, Hou J, Chen X, Wang L and Wu X: Unveiling the role of UPF3B in hepatocellular carcinoma: Potential therapeutic target. Cancer Sci. 115:2646–2658. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Wang H, Qian D, Wang J, Liu Y, Luo W, Zhang H, Cheng J, Li H, Wu Y, Li W, et al: HnRNPR-mediated UPF3B mRNA splicing drives hepatocellular carcinoma metastasis. J Adv Res. 68:257–270. 2025. View Article : Google Scholar : | |
|
Ma J, Chen T, Wu S, Yang C, Bai M, Shu K, Li K, Zhang G, Jin Z, He F, et al: iProX: An integrated proteome resource. Nucleic Acids Res. 47:D1211–D121. 2019. View Article : Google Scholar : | |
|
Chen T, Ma J, Liu Y, Chen Z, Xiao N, Lu Y, Fu Y, Yang C, Li M, Wu S, et al: iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 50:D1522–D1527. 2022. View Article : Google Scholar : |










