



Impact of the ATM/Chk2 pathway and cell cycle phase on radiation‑induced senescence in A549 human lung cancer cells
- Authors:
- Published online on: August 26, 2025 https://doi.org/10.3892/br.2025.2047
- Article Number: 169
-
Copyright: © Sato et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Radiation therapy is one of the main treatment options for various cancers, including lung cancer (1,2). Ionizing radiation induces biological effects through DNA damage, and the DNA damage response (DDR) is associated with cellular responses to radiation, such as DNA repair, apoptosis and cellular senescence (3-5). Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related (ATR) are master regulators of DDR (6). ATM and ATR are involved in the regulation of the response to DNA double-strand breaks (DSBs) and single-strand breaks (SSBs), respectively (6-8). These molecules activate downstream molecules such as checkpoint kinases (Chks) and promote DNA repair by temporarily arresting the cell cycle (7,9). The cell cycle contributes to the cellular response to radiation. For example, the radiosensitivity of cells varies depending on the cell cycle stage at the time of irradiation (10). Furthermore, the selection of the DSB repair pathway depends on the cell cycle stage. In brief, homologous recombination repair functions only in the late S and G2 phases, whereas non-homologous end-joining occurs throughout the cell cycle, excluding M phase (9).
Cellular senescence is a state of stable proliferation arrest induced by DNA damage, including that induced by ionizing radiation. Senescent cells are characterized by morphological changes such as enlargement and flattening, high expression of cell cycle regulators such as p21, high activity of senescence-associated β-galactosidase (SA-β-gal), and a senescence-associated secretory phenotype (SASP), which secretes factors such as inflammatory factors (11-13). Senescence is considered a cancer suppression mechanism.
The induction of senescence in cancer cells exhibiting unrestricted proliferation is beneficial. Induction of senescence by chemotherapy and radiation therapy contributes to tumor suppression (14,15). However, cancer therapy can induce cell senescence, and the resulting SASP causes inflammation in surrounding tissue, leading to harmful effects such as tissue damage, age-related diseases, and tumor promotion (16,17). To improve the adverse effects of senescent cells, senolytic agents, which eliminate senescent cells, have attracted attention recently (18,19). For example, the removal of senescent cells by the senolytic agent ABT-263 improves harmful effects such as hematopoietic deterioration following total-body irradiation and the impairment of cardiac function (20-22). Therefore, it is hypothesized that the application of senolytic agents may improve the efficacy of cancer therapy, including radiation therapy. However, not all irradiated cancer cells exhibit characteristics of senescence (23). This may be undesirable for radiation therapy because these cells may maintain proliferation potential and show low response to senolytic agents (23,24). Therefore, inducing senescence in irradiated cells may improve the efficacy of the combination of cancer therapy and senolytic agents (25,26). However, understanding the mechanism controlling the induction of senescence in cancer cells by cancer therapy, including radiation therapy, is insufficient. Therefore, it is necessary to clarify the mechanism and role of cell senescence induction.
As aforementioned, DDR and the cell cycle are associated with cell responses to radiation; therefore, the present study aimed to explore the factors controlling radiation-induced senescence in A549 human lung cancer cells, focusing on DDR factors and the cell cycle.
Materials and methods
Reagents
Dulbecco's PBS (-; Ca2+- and Mg2+-free) and thymidine (cat. no. 207-19421) were purchased from Wako Pure Chemical Industries, Ltd. Propidium iodide (PI), DMSO, caffeine, nutlin-3α (cat. no. N6287), Chk2 inhibitor II (cat. no. 220486) and pifithrin-α (cat. no. 506132) were purchased from Sigma-Aldrich (Merck KGaA). Senescence β-Galactosidase Activity Assay kit (cat. no. #35302), anti-phosphorylated (p-)Chk2 (Thr68; cat. no. #2661), anti-Chk2 (cat. no. #3440), anti-p-histone H2A.X (γH2AX; cat. no. #9718), anti-H2A.X (cat. no. #2595), anti-p21 (cat. no. #2947), anti-p53 (cat. no. #9282), anti-GAPDH (cat. no. #5174) and anti-mouse (cat. no. #7076) and anti-rabbit IgG HRP-linked antibody (cat. no. #7074) were purchased from Cell Signaling Technology Japan, K.K. Chk1 inhibitor SCH900776 was purchased from MedChemExpress. Olaparib (cat. no. S1060) was purchased from Selleck Chemicals.
Cell culture and treatment
The human lung adenocarcinoma cell line A549 was obtained from the RIKEN Bio-Resource Research Center (Tsukuba, Japan). A549 cells were maintained in low-glucose DMEM (Wako Pure Chemical Industries) supplemented with 10% heat-inactivated FBS (Sigma-Aldrich; Merck KGaA) and 1% penicillin/streptomycin (Wako Pure Chemical Industries). Cells were cultured at 37˚C in a humidified atmosphere of 5% CO2/95% air.
Cells (1.0x105) were seeded in 35-mm culture dishes (Sumitomo Bakelite Co., Ltd.) and incubated at 37˚C in a humidified atmosphere of 5% CO2/95% air for 6 h to allow adherence. Inhibitors were added to the culture medium and incubated for 1 h before X-ray irradiation. The inhibitors used were as follows: caffeine (2 mM) for ATM/ATR, SCH900776 (400 nM) for Chk1, Chk2 inhibitor II (10 µM) for Chk2, nutlin-3α (10 µM) for mouse double minute protein 2 (MDM2), pifithrin-α (10 µM) for p53, and olaparib (1 µM) for poly ADP-ribose polymerase (PARP). Following drug treatment, cells were irradiated with X-rays at room temperature using an X-ray generator (MBR-1520R-3; Hitachi Medical Corporation) at 150 kVp and 20 mA with 0.5-mm aluminum and 0.3-mm copper filters. The distance from the X-ray source to the sample was 45 cm, and the dose rate was 0.99-1.02 Gy/min. After irradiation, the cells were cultured at 37˚C for 0.5 h to 4 days and then harvested using 0.25% trypsin-ethylenediaminetetraacetic acid (Wako Pure Chemical Industries) for subsequent experiments.
Small interfering (si)RNA transfection
Chk2 knockdown was performed using Silencer® Select Pre-designed siRNA targeting Chk2 (cat. no. s533978) and p53 (siRNA#1, cat. no. s606; siRNA#2, cat. no. s607) and RNAiMAX (all Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Transfection was performed at 37˚C in a humidified atmosphere of 5% CO2/95% air. The forward sequences for Chk2 was 5'-CUCUUACAUUGCAUACAUATT-3' (reverse sequences: 5'-UAUGUAUGCAAUGUAAGAGTT-3'). The forward sequences for p53 #1 and #2 were 5'-GAAAUUUGCGUGUGGAGUATT-3' (reverse sequences: 5'-UACUCCACACGCAAAUUUCCT-3') and 5'-GGUGAACCUUAGUACCUAATT' (reverse sequences: 5'-UUAGGUACUAAGGUUCACCAA-3'), respectively. Silencer® Select Negative Control #1 (cat. no. 4390843, Thermo Fisher Scientific, Inc., sequence not available) was used as the negative control. The final concentration of siRNA was 10 nM. After 48 h transfection, cells were harvested and immediately subjected to subsequent experiments.
Analysis of SA-β-gal activity and cell size
SA-β-gal activity was analyzed using the Senescence β-Galactosidase Activity Assay kit according to the manufacturer's instructions. In brief, after irradiation, the culture medium was removed and the cells were treated with bafilomycin A1 (100 nM) for 1 h at 37˚C. SA-β-Gal Substrate Solution (33 µM) was added at 37˚C for 2 h. After washing with PBS, the cells were harvested and washed again with PBS. The cells were suspended in cold PBS containing 2% FBS, and the fluorescence intensity of SA-β-Gal Substrate was analyzed using a flow cytometer (CytoFLEX with CytExpert software version 2.4.0.28; Beckman-Coulter, Inc.). The cell size was estimated by the forward scatter signal; the intensity correlates with the relative size of cells.
Cell cycle analysis
Cell cycle analysis was performed as previously reported (27). Harvested cells were fixed overnight in ice-cold 70% ethanol at -20˚C. The fixed cells were washed with PBS and then treated with RNase (200 µg/ml) at 37˚C for 30 min. Cells were resuspended in PBS containing PI and incubated at 37˚C in the dark for 30 min. The cells were passed through a cell strainer (BD Falcon; BD Biosciences) and analyzed using a flow cytometer (CytoFLEX with CytExpert software version 2.4.0.28; Beckman-Coulter, Inc.).
SDS-PAGE and western blotting
SDS-PAGE analysis and western blotting were performed as previously reported (28). The following primary antibodies were used: Anti-p-Chk2, anti-Chk2, anti-γH2AX, anti-H2AX, anti-p53 antibody, anti-p21 (all 1:3,000) and anti-GAPDH (1:4,000). HRP-labeled anti-rabbit IgG was used as a secondary antibody (1:10,000). Antigens were visualized using Clarity™ Western ECL Substrate (Bio-Rad Laboratories Inc.). Stripping Solution (Wako Pure Chemical Industries) was used for blot stripping. GAPDH was used as a loading control.
Cell cycle synchronization
Cell cycle synchronization was performed by the double thymine block method, as previously reported, with modifications (29). In brief, cells were cultured in low-glucose DMEM (Wako Pure Chemical Industries) supplemented with thymidine (2 mM) at 37˚C for 18 h. Subsequently, cells were washed with thymidine-free culture medium and incubated for 9 h at 37˚C. Then, fresh culture medium containing thymidine (2 mM) was added, followed by culture at 37˚C for 18 h. After washing with prewarmed culture medium, cells synchronized at the G1/S boundary (prior to DNA synthesis) were incubated with fresh culture medium to allow cell cycle progression. The G1/S synchronization was confirmed by cell cycle analysis.
Statistical analysis
All data are presented as the mean ± standard deviation of at least three independent experiments. Comparisons were performed via unpaired two-sided Student's t- or the Mann-Whitney U-test depending on the data distribution using Excel 2016 software (Microsoft Corporation) along with Statcel 4 add-in software (The Publisher OMS Ltd.). One-way ANOVA followed by Tukey-Kramer post hoc test was used for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
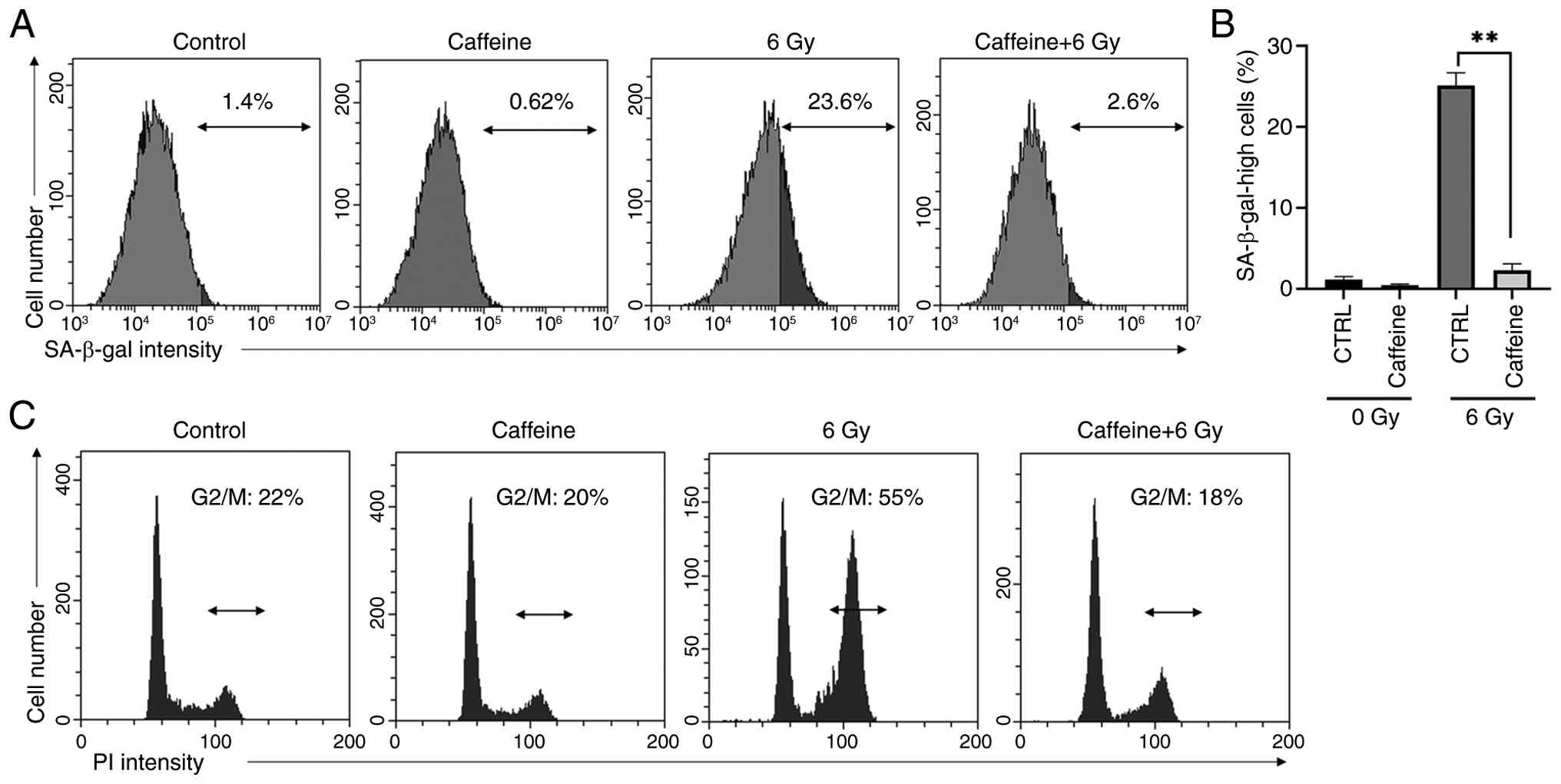
ATM/ATR inhibitor caffeine suppresses radiation-induced cell senescence and cell cycle arrest
The proportion of cells with high SA-β-gal activity following irradiation was reduced by the ATM/ATR inhibitor caffeine (Fig. 1A and B). Furthermore, caffeine suppressed radiation-induced G2/M arrest in irradiated A549 cells (Fig. 1C).
Chk2 is involved in radiation-induced cell senescence
As aforementioned, caffeine suppressed radiation-induced cell senescence and G2/M arrest, implying a relationship between radiation-induced cell senescence and G2/M arrest. Therefore, the present study investigated the involvement of G2/M arrest in radiation-induced senescence using inhibitors of Chk1 and Chk2, which are downstream molecules of ATR and ATM, respectively. Chk1 inhibitor, but not the Chk2 inhibitor, suppressed radiation-induced G2/M arrest (Fig. 2A). Of note, the Chk2 inhibitor decreased the number of senescent cells with high radiation-induced SA-β-gal activity, whereas the Chk1 inhibitor had no effect (Fig. 2B and C). In addition, western blot analysis confirmed that the Chk2 inhibitor decreased the phosphorylation of Chk2 in the 6 Gy-irradiated cells (Fig. 2D). These results suggested the involvement of Chk2, but not Chk1, in the induction of cellular senescence by radiation. In addition, knockdown of Chk2 resulted in the decrease in senescent cells with high radiation-induced SA-β-gal activity (Fig. 2E and F).
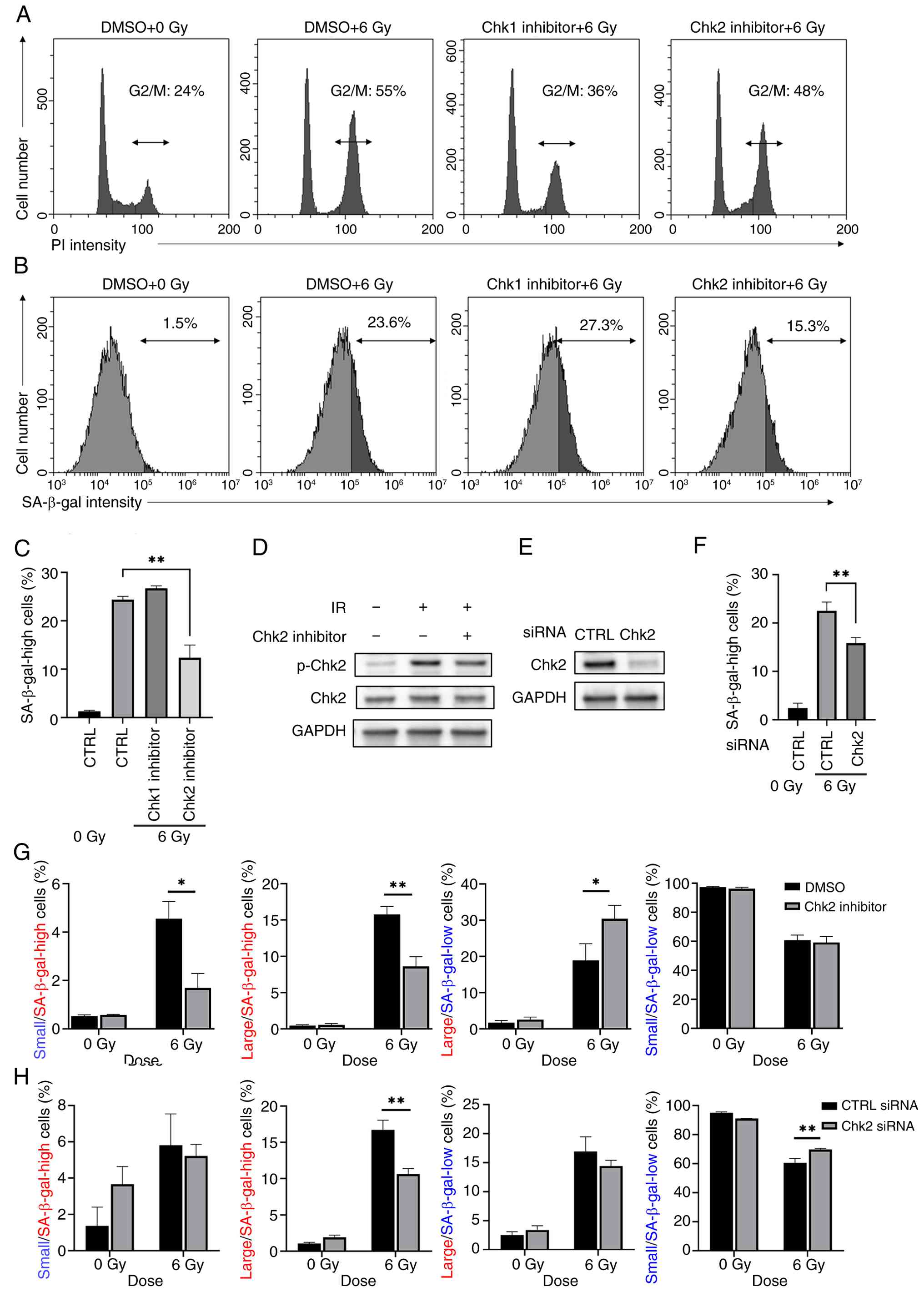
Because the radiation-induced senescent cancer cell population is heterogeneous (23), the present study analyzed the effect of Chk2 inhibition on the proportion of A549 cells exhibiting senescent characteristics, including high SA-β-gal activity and increased cell size. Chk2 inhibitor decreased the proportion of large and small cells with high SA-β-gal activity, whereas the proportion of large cells with low SA-β-gal activity was increased (Fig. 2G). Knockdown of Chk2 also decreased the percentage of large cells with high SA-β-gal activity (Fig. 2H). However, in contrast to Chk2 inhibitor, no increase in the proportion of large cells with low SA-β-gal activity was observed following Chk2 knockdown, while it increased the percentage of small cells with low SA-β-gal activity (Fig. 2H).
Olaparib enhances radiation-induced senescence in A549 cells
Because the ATM/Chk2 pathway is activated by DSBs (6), it was hypothesized that DSBs are important for the induction of radiation-induced senescence in A549 cells. Impairment of SSBs repair leads to DSBs (30). Therefore, it was hypothesized that the inhibition of SSBs repair may enhance radiation-induced cell senescence. The present study analyzed the effect of olaparib, which inhibits the SSB repair factor PARP and converts SSBs into DSBs (31), on radiation-induced senescence in A549 cells. Combination of olaparib with radiation effectively increased the radiation-induced γH2AX expression (Fig. 3A), which is a representative DSB marker (32). Olaparib did not increase the number of senescent A549 cells with high SA-β-gal activity in the absence of radiation (Fig. 3B and C). However, olaparib significantly increased the number of radiation-induced senescent A549 cells with high SA-β-gal activity (Fig. 3C). In addition, the effect of olaparib on radiation-induced senescence was confirmed by the upregulation of p21 expression, a senescence marker (Fig. 3D) (33). Western blot analysis demonstrated that p-Chk2 and p53 were markedly upregulated in the olaparib-treated group following irradiation (Fig. 3D). These results suggested that inhibition of SSB repair by olaparib leads to enhanced activation of the ATM/Chk2/p53 pathway, thereby promoting radiation-induced senescence in A549 cells.
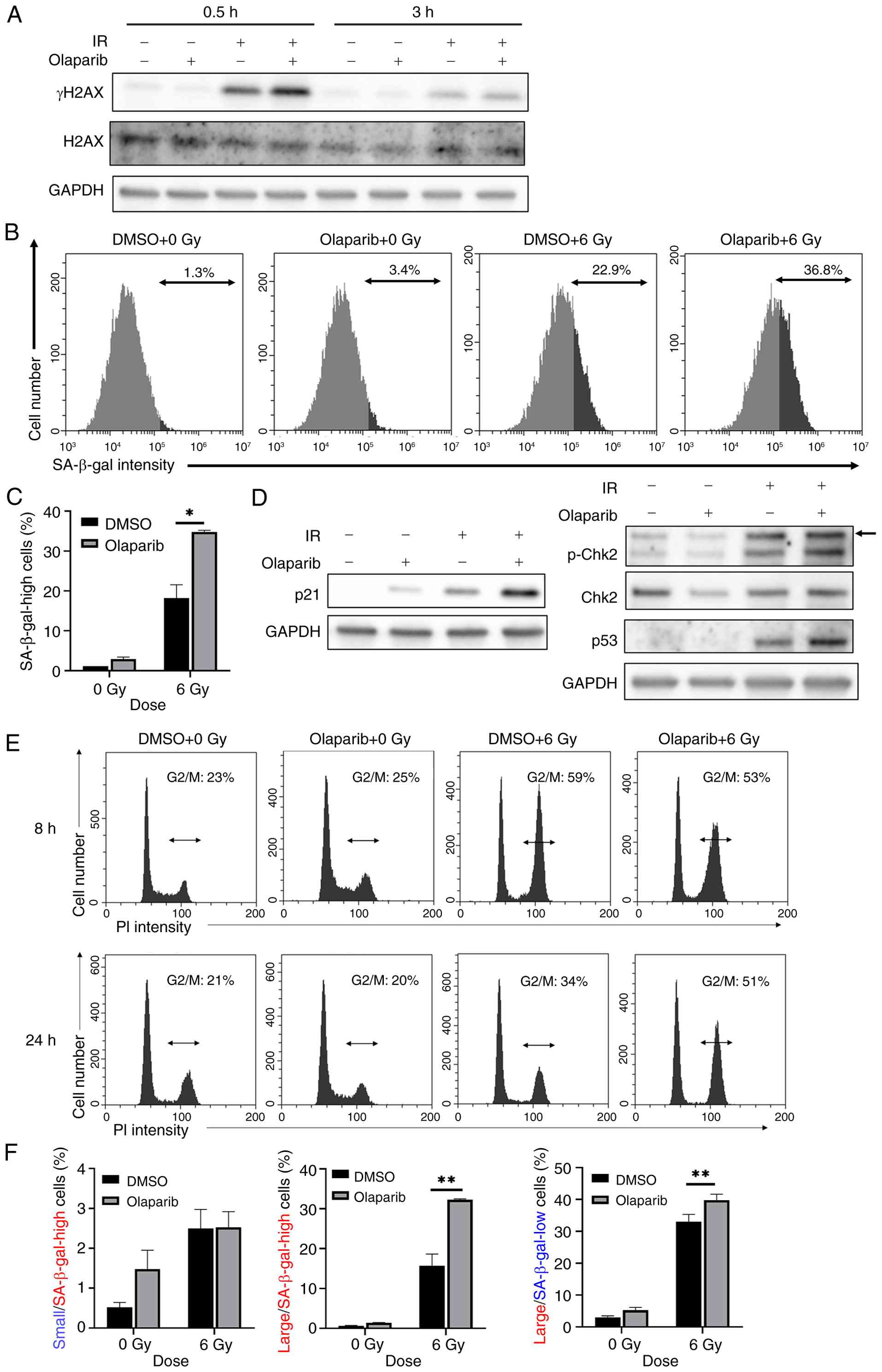
The present study examined the effect of olaparib on cell cycle distribution following irradiation. At 8 h post-irradiation, no apparent differences were observed in the G2/M population between the olaparib-treated and control groups. However, at 24 h post-irradiation, a notable increase in the G2/M population was observed in olaparib-treated cells (Fig. 3E), suggesting that the combination of olaparib and radiation promotes sustained G2/M arrest. Olaparib increased the proportion of large cells with both high and low SA-β-gal activity compared with the control (Fig. 3F).
Nutlin-3α promotes senescence in irradiated A549 cells
The ATM/Chk2 pathway activates p53, which can induce cell senescence (34). Knockdown of p53 or p53 inhibitor pifithrin-α partly decreased radiation-induced senescence in A549 cells (Fig. 4A-C), suggesting radiation induced senescence in A549 cells through p53. Therefore, it was hypothesized that the ATM/Chk2/p53 pathway could be a potential target for the induction of senescence in A549 cells. The present study investigated whether a pharmaceutical activator of p53 enhances radiation-induced senescence; nutlin-3α inhibits MDM2, thereby increasing p53 expression (35). Nutlin-3α increased the protein expression of p53 to the same extent as 6 Gy radiation, and their combination further increased p53 expression compared with the effect of each stimulus each alone (Fig. 4D). Although nutlin-3α increased the proportion of cells with high SA-β-gal activity following irradiation, similar to the effects of olaparib, nutlin-3α alone also increased the proportion of cells with high SA-β-gal activity to a similar extent as radiation alone (Fig. 4E and F). Western blot analysis demonstrated that nutlin-3α increased p21 expression following irradiation (Fig. 4D), consistent with activation of the p53/p21 axis. By contrast, no increase in p-Chk2 was observed in nutlin-3α-treated cells (Fig. 4D), suggesting that nutlin-3α enhanced radiation-induced senescence through a pathway independent of Chk2 activation.
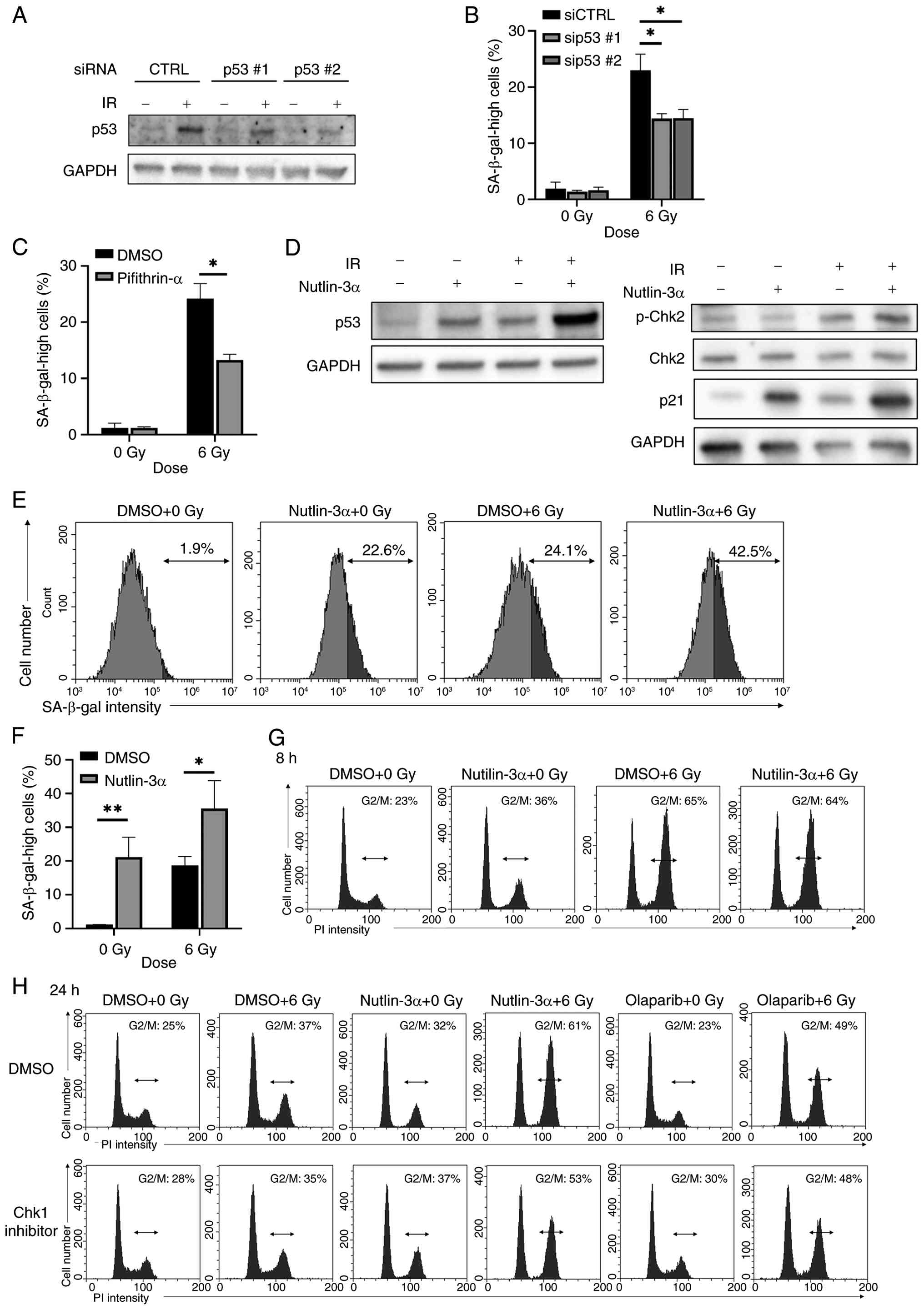
To investigate the effects of nutlin-3α, the present study analyzed cell cycle distribution following irradiation. Nutlin-3α did not affect the G2/M population at 8 h post-irradiation (Fig. 4G), but markedly increased the G2/M population at 24 h (Fig. 4H), similar to the effect observed with olaparib. This increase in the G2/M population induced by either nutlin-3α or olaparib was also observed in the presence of Chk1 inhibitor, indicating a Chk1-independent mechanism.
Effect of the cell cycle phase on radiation-induced cell senescence
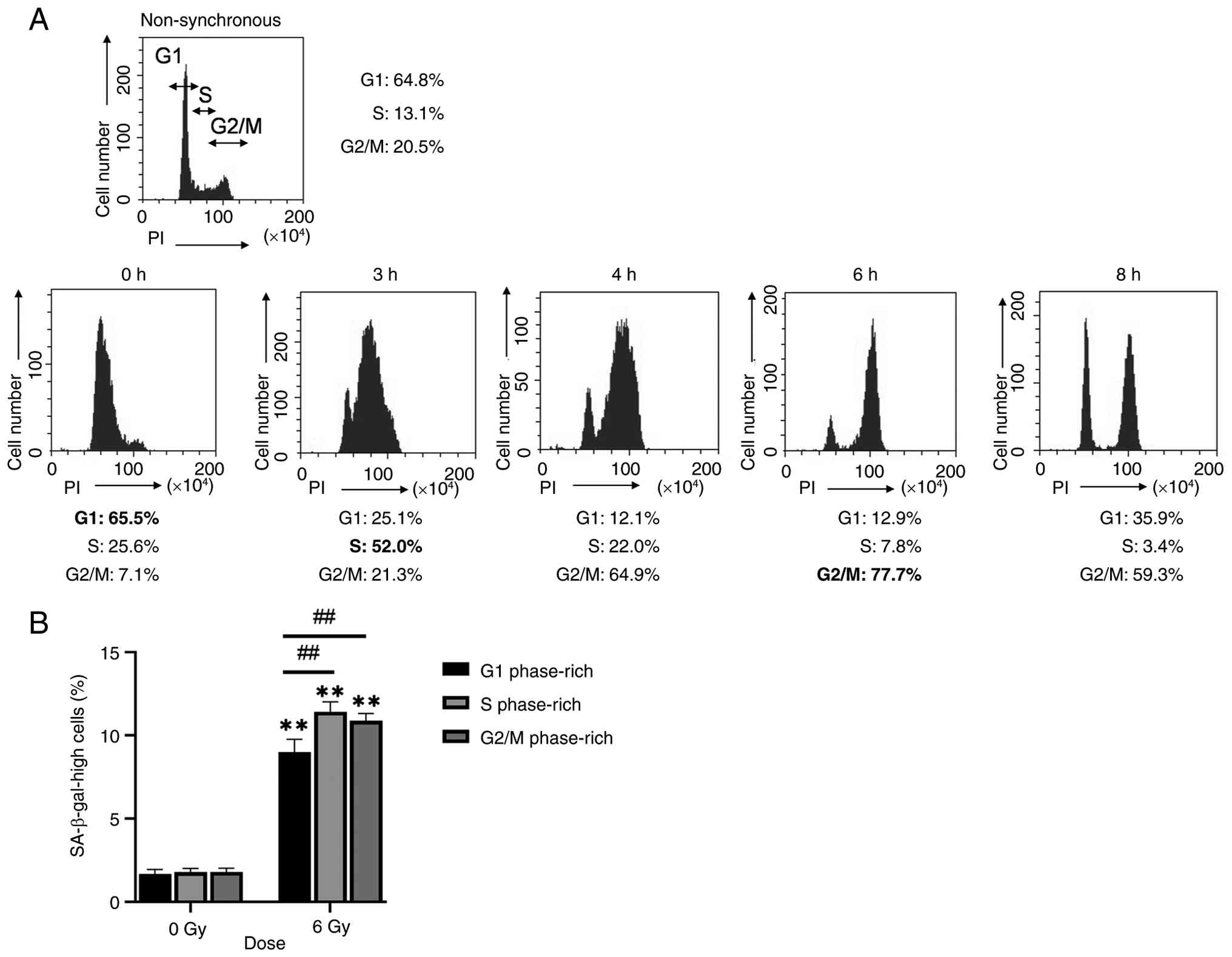
The present study investigated the association between the cell cycle phase at the time of irradiation and radiation-induced cell senescence. The present study determined the time points at which cells predominantly entered each phase of the cell cycle following release from synchronization. The highest percentages of cells in G1, S and G2/M phases were obtained at 0, 3 and 6 h after release, respectively (Fig. 5A). Therefore, X-ray irradiation was performed at these time points. The proportion of cells with high SA-β-gal activity was significantly higher following irradiation during S phase G2/M phase than during G1 phase (Fig. 5B). These results suggested that the cell cycle stage during irradiation affects radiation-induced cellular senescence.
Discussion
Cell senescence arrests proliferation of cancer cells (36), which contributes tumor suppression. A negative effect of senescent cells is the production of SASP, which can promote cancer progression and tissue damage. For example, in lung cancer cells, although anti-cancer drug palbociclib induces senescence and inhibits the proliferation of cancer cells, SASP promotes the migration and invasion of cancer cells (37). In addition, SASP causes tissue damage such as pulmonary fibrosis (38). However, these negative effects may be improved by senolytic agents (39). Promoting radiation-induced senescence in cancer cells and minimizing the negative effects leads to an improvement in outcomes following radiotherapy for lung cancer.
To identify potential targets that regulate radiation-induced senescence in cancer cells, the present study focused on DDR factors and the cell cycle and their association with radiation-induced cell senescence. The present study revealed the involvement of the ATM/Chk2/p53 pathway in the induction of senescence by radiation in A549 cells. In addition, olaparib or radiation irradiation during specific cell cycle phases resulted in the enhancement of radiation-induced senescence in A549 cells, suggesting the ATM/Chk2/p53 pathway and the cell cycle phase at the time of irradiation are important for controlling radiation-induced senescence in cancer cells.
Following DNA damage, Chk2 is phosphorylated by ATM (40) and activates downstream signaling, leading to DDR and cell cycle arrest. Here, Chk2 inhibitor II, an ATP competitive Chk2 kinase inhibitor (41), decreased the proportion of senescent cells, as well as the phosphorylation of Chk2 in the irradiated cells, suggesting the involvement of Chk2 in radiation-induced senescence in A549 cells. The role of the ATM/Chk2 pathway in DNA damage-induced senescence has been demonstrated by various studies (42-45). In addition, the pathway is involved in replicative senescence of normal human fibroblasts (46). As cell senescence in normal lung tissue leads to undesirable effects such as pulmonary fibrosis following radiation therapy (38), the ATM/Chk2 pathway may be a promising therapeutic target for decreasing radiation-induced damage in normal tissue while simultaneously enhancing the efficacy of cancer treatment.
Although both Chk2 inhibitor and Chk2 knockdown decreased the proportion of cells with high SA-β-gal activity, Chk2 inhibitor decreased the proportion of small cells with high SA-β-gal activity but increased the population of large cells with low SA-β-gal activity, while Chk2 knockdown did not. Notably, although Chk2 inhibition decreased the proportion of cells with high SA-β-gal activity, it also led to an increase in small cells with high SA-β-gal activity. As senescent cells are typically enlarged, this subpopulation may reflect cells with elevated lysosomal activity that do not fully correspond to classical senescence. It has been reported that during cell senescence, morphological changes (early state, characterized by cell enlargement and flattening) occur before SA-β-gal activity is elevated (fully established senescence) (47). Therefore, Chk2 phosphorylation is hypothesized to be involved in the transition from the early to the late state of senescence. Chk2 inhibitor may affect other processes, such as cell cycle progression, DNA repair, or apoptotic signaling, potentially reflecting off-target or non-specific effects of the inhibitor that are not caused by siRNA-mediated knockdown. Furthermore, compensatory mechanisms involving other Chks such as Chk1 or DDR pathways may partially counteract the effects of Chk2 knockdown (48,49), resulting in an incomplete decrease in senescent subpopulations. This hypothesis is supported by the present observation that caffeine almost completely abolished radiation-induced senescence. However, both approaches have inherent technical limitations: Chemical inhibitors may vary in specificity and intracellular distribution, while siRNA-mediated knockdown is transient and may leave residual protein activity sufficient to maintain partial function. Future studies using genetic knockout models (clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9) and alternative inhibitors are needed to distinguish Chk2-specific effects from potential off-target or compensatory pathways.
p53 knockdown or pifithrin-α decreased radiation-induced senescence by ~10%. These results suggested that radiation induced senescence in A549 cells predominantly through p53. However, as senescence was not completely abrogated by p53 knockdown, additional p53-independent mechanisms may also contribute to the senescence phenotype. These may include alternative effectors such as p16 (33,50). Additionally, incomplete knockdown efficiency of p53 by siRNA may have contributed to the remaining senescent population. To elucidate the contribution of these pathways, studies using complete gene knockout models are necessary.
Because the ATM/Chk2 pathway is activated in response to DSBs, leading to p53 activation (34,51), the present study investigated whether modulators of DSBs or p53 affect radiation-induced senescence. Olaparib induces DSBs by inhibiting SSB repair via PARP inhibition (31); combination of olaparib and radiation effectively increased the number of senescent A549 cells. Similarly, Huart et al (52) recently reported that olaparib promotes radiation-induced cell senescence. Taken together, these findings suggest that strategies to increase DSBs in irradiated cells may be useful for enhancing cell senescence.
p53 knockdown decreased radiation-induced senescence by ~10%. This reduction was similar to that following Chk2 knockdown. Additionally, treatment with olaparib prior to irradiation increased the number of senescent cells compared with radiation alone, accompanied by elevated levels of p-Chk2 and p53 protein. Consistent with these findings, cell cycle analysis showed that olaparib did not decrease G2/M arrest immediately following irradiation but increased the G2/M population at 24 h post-irradiation, suggesting that olaparib may delay progression through G2/M phase, thereby enhancing the accumulation of DSBs and activation of the ATM-Chk2-p53 axis. These results support the hypothesis that activation of the Chk2-p53 axis contributes to the enhancement of radiation-induced cell senescence. Taken together, these findings indicate that Chk2 primarily regulated radiation-induced cell senescence through p53, although other pathways may also be involved.
The combination of the p53 activator nutlin-3α and radiation effectively increased the number of senescent cells compared with each treatment alone. Of note, the combination increased p53 and p21 expression without elevating p-Chk2 levels, highlighting the role of the p53/p21 pathway in nutlin-3α-mediated senescence. Furthermore, cell cycle analysis revealed that nutlin-3α increased the G2/M population at 24 h post-irradiation, and this increase was not suppressed by co-treatment with Chk1 inhibitor, suggesting nutlin-3α promoted G2/M arrest through a Chk1-independent mechanism, potentially driven by p53/p21 activation (53,54). A similar Chk1-independent increase in G2/M arrest was observed in olaparib-treated irradiated cells. Together, these results suggested that Chk1-independent but p53-dependent G2/M arrest may contribute to radiation-induced senescence, whereas Chk1-dependent G2/M arrest did not.
The proportion of senescent cells following the combination of the p53 activator nutlin-3α and radiation was comparable with that of cells treated with olaparib and radiation. This suggested that olaparib and nutlin-3α enhanced radiation-induced senescence via the p53 pathway. However, nutlin-3α, but not olaparib, induced senescence in non-irradiated cells. This indicated that olaparib selectively enhanced radiation-induced senescence in cancer cells without inducing senescence in non-irradiated normal cells, emphasizing the potential usefulness of olaparib compared with p53 activators for radiation therapy.
There are clinical studies of olaparib for lung cancer, but olaparib maintenance therapy does not improve progression-free survival (55,56). However, certain reports have demonstrated that combined olaparib and radiation effectively induces antitumor effects for lung cancer by increasing radiosensitivity of tumors in vitro and in vivo (57,58), suggesting the use of combined olaparib and radiotherapy for lung cancer in clinical oncology settings. Here, olaparib effectively increased radiation-induced senescence in lung cancer A549 cells. The regulation of cell senescence by olaparib may contribute to the effects of combination therapy.
The analysis of populations classified by senescent features in olaparib-treated irradiated A549 cells revealed that the combination of olaparib and radiation increased both the large SA-β-gal-high and -low activity cell populations. Because our previous study suggested that large SA-β-gal-low activity cells might contribute to the survival of irradiated A549 cells (23), the population stimulated by the combination of olaparib and irradiation may be an obstacle to the efficacy of radiation therapy. Therefore, this cell population needs to be regulated to improve the efficacy of radiation and olaparib.
Although the present study primarily aimed to elucidate the molecular mechanisms underlying radiation-induced cell senescence and its modulation by DDR, the SASP represents a key aspect of the senescence response, particularly in shaping the tissue microenvironment and influencing therapy outcomes (59,60). Due to resource limitations, SASP profiling was not performed in the present study. Future studies should assess key inflammatory mediators such as IL-6 and -8 through transcriptomic and/or proteomic approaches to clarify the biological impact of senescent cells induced by combined radiation and olaparib.
The cell cycle phase at the time of irradiation (S or G2/M phase) influenced radiation-induced senescence. This is in line with the findings of Hsu et al (61), who reported that the dynamics of p21 determine the proliferation/senescence cell fate following chemotherapy and this fate is associated with the cell cycle phase during treatment. In brief, the aforementioned study reported that a small proportion of senescence-fated cells express high levels of p21 during treatment, whereas most senescence-fated cells have delayed p21 responses. Of note, high p21 levels are observed in G1 phase, whereas delayed responses were associated with S/G2 phase. These results demonstrated that the mechanisms by which chemotherapy induces senescence vary depending on the cell cycle state during drug treatment and drug-induced senescence is more likely to occur during treatment in S/G2 phase. As changes in the cell cycle phase following irradiation vary depending on the cell cycle phase at irradiation (62), the cell cycle kinetics following irradiation may be associated with radiation-induced senescence.
Radiation-induced DSBs are more frequent in G2/M than in G1 phase cells, potentially due to the doubled DNA content present during G2/M. A comet assay-based analysis of ataxia telangiectasia lymphoblasts has revealed that G2/M phase cells exhibited extensive DNA damage compared with G0/G1 phase cells following irradiation (63). These findings support the hypothesis that the lower proportion of SA-β-gal-positive cells observed after G1 phase irradiation may be due to decreased levels of DNA damage, which is consistent with the lower DNA content of G1 phase cells.
In conclusion, the present results demonstrated that the ATM/Chk2 pathway and the DNA content at the time of irradiation are involved in radiation-induced senescence in A549 cells, and the regulation of Chk2 activity and cell cycle phase at irradiation enhances radiation-induced senescence. Although further studies, including validation of the present results in vivo, are needed, enhancing radiation-induced senescence by targeting these factors may complement the efficacy of the combination of radiation therapy and senolytic agents.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Interdisciplinary Collaborative Research Grant for Young Scientists, Hirosaki University, JSPS KAKENHI (grant no. JP21K07691) and Grant for Scientific Research in Master Course in Hirosaki University Graduate School of Health Sciences.
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
HY conceived the study, designed the methodology, analyzed data and wrote the manuscript. KS, YS, FS, NM and ET analyzed data. KS wrote the manuscript. ET interpreted data and edited the manuscript. All authors have read and approved the final manuscript. KS and HY confirm the authenticity of all the raw data.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Schaue D and McBride WH: Opportunities and challenges of radiotherapy for treating cancer. Nat Rev Clin Oncol. 12:527–540. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Toulany M: Targeting DNA double-strand break repair pathways to improve radiotherapy response. Genes (Basel). 10(25)2019.PubMed/NCBI View Article : Google Scholar | |
|
Wilkinson B, Hill MA and Parsons JL: The cellular response to complex DNA damage induced by ionising radiation. Int J Mol Sci. 24(4920)2023.PubMed/NCBI View Article : Google Scholar | |
|
Khan MGM and Wang Y: Advances in the current understanding of how low-dose radiation affects the cell cycle. Cells. 11(356)2022.PubMed/NCBI View Article : Google Scholar | |
|
Balagurumoorthy P, Adelstein SJ and Kassis AI: Novel method for quantifying radiation-induced single-strand-break yields in plasmid DNA highlights 10-fold discrepancy. Anal Biochem. 417:242–246. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Weber AM and Ryan AJ: ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 149:124–138. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Li S, Wang L, Wang Y, Zhang C, Hong Z and Han Z: The synthetic lethality of targeting cell cycle checkpoints and PARPs in cancer treatment. J Hematol Oncol. 15(147)2022.PubMed/NCBI View Article : Google Scholar | |
|
Rundle S, Bradbury A, Drew Y and Curtin NJ: Targeting the ATR-CHK1 axis in cancer therapy. Cancers (Basel). 9(41)2017.PubMed/NCBI View Article : Google Scholar | |
|
Sia J, Szmyd R, Hau E and Gee HE: Molecular mechanisms of radiation-induced cancer cell death: A primer. Front Cell Dev Biol. 8(41)2020.PubMed/NCBI View Article : Google Scholar | |
|
Shimono H, Kaida A, Homma H, Nojima H, Onozato Y, Harada H and Miura M: Fluctuation in radioresponse of HeLa cells during the cell cycle evaluated based on micronucleus frequency. Sci Rep. 10(20873)2020.PubMed/NCBI View Article : Google Scholar | |
|
Sikora E, Bielak-Żmijewska A and Mosieniak G: What is and what is not cell senescence. Postepy Biochem. 64:110–118. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Ogrodnik M: Cellular aging beyond cellular senescence: Markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell. 20(e13338)2021.PubMed/NCBI View Article : Google Scholar | |
|
Birch J and Gil J: Senescence and the SASP: Many therapeutic avenues. Genes Dev. 34:1565–1576. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Ewald JA, Desotelle JA, Wilding G and Jarrard DF: Therapy-induced senescence in cancer. J Natl Cancer Inst. 102:1536–1546. 2010.PubMed/NCBI View Article : Google Scholar | |
|
Park B, Yee C and Lee KM: The effect of radiation on the immune response to cancers. Int J Mol Sci. 15:927–943. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Peng X, Wu Y, Brouwer U, van Vliet T, Wang B, Demaria M, Barazzuol L and Coppes RP: Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche. Cell Death Dis. 11(854)2020.PubMed/NCBI View Article : Google Scholar | |
|
Tripathi U, Misra A, Tchkonia T and Kirkland JL: Impact of senescent cell subtypes on tissue dysfunction and repair: Importance and research questions. Mech Ageing Dev. 198(111548)2021.PubMed/NCBI View Article : Google Scholar | |
|
Saliev T and Singh PB: Targeting senescence: A review of senolytics and senomorphics in anti-aging interventions. Biomolecules. 15(860)2025.PubMed/NCBI View Article : Google Scholar | |
|
Czajkowski K, Herbet M, Murias M and Piątkowska-Chmiel I: Senolytics: Charting a new course or enhancing existing anti-tumor therapies? Cell Oncol (Dordr). 48:351–371. 2025.PubMed/NCBI View Article : Google Scholar | |
|
Kim JH, Brown SL and Gordon MN: Radiation-induced senescence: Therapeutic opportunities. Radiat Oncol. 18(10)2023.PubMed/NCBI View Article : Google Scholar | |
|
Lérida-Viso A, Estepa-Fernández A, Morellá-Aucejo Á, Lozano-Torres B, Alfonso M, Blandez JF, Bisbal V, Sepúlveda P, García-Fernández A, Orzáez M and Martínez-Máñez R: Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol Res. 183(106356)2022.PubMed/NCBI View Article : Google Scholar | |
|
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al: Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 22:78–83. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Sato K, Iwasaki S and Yoshino H: Effects and related mechanisms of the senolytic agent ABT-263 on the survival of irradiated A549 and Ca9-22 cancer cells. Int J Mol Sci. 22(13233)2021.PubMed/NCBI View Article : Google Scholar | |
|
Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al: Senescence-associated reprogramming promotes cancer stemness. Nature. 553:96–100. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al: The Achilles' heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell. 14:644–658. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, Wolter K, Pombo J, Irvine EE, Innes AJ, et al: Cardiac glycosides are broad-spectrum senolytics. Nat Metab. 1:1074–1088. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Wakasaya T, Yoshino H, Fukushi Y, Yoshizawa A and Kashiwakura I: A liquid crystal-related compound induces cell cycle arrest at the G2/M phase and apoptosis in the A549 human non-small cell lung cancer cell line. Int J Oncol. 42:1205–1211. 2013.PubMed/NCBI View Article : Google Scholar | |
|
Yoshino H, Kumai Y and Kashiwakura I: Effects of endoplasmic reticulum stress on apoptosis induction in radioresistant macrophages. Mol Med Rep. 15:2867–2872. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Chen G and Deng X: Cell synchronization by double thymidine block. Bio Protoc. 8(e2994)2018.PubMed/NCBI View Article : Google Scholar | |
|
Chatterjee N and Walker GC: Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 58:235–263. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, Cruz C, Oaknin A, Kaye SB and de Bono JS: A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 30:1437–1447. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Kuo LJ and Yang LX: Gamma-H2AX-a novel biomarker for DNA double-strand breaks. In Vivo. 22:305–309. 2008.PubMed/NCBI | |
|
Serrano M, Lin AW, McCurrach ME, Beach D and Lowe SW: Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 88:593–602. 1997.PubMed/NCBI View Article : Google Scholar | |
|
Nagasaka M, Hashimoto R, Inoue Y, Ishiuchi K, Matsuno M, Itoh Y, Tokugawa M, Ohoka N, Morishita D, Mizukami H, et al: Anti-tumorigenic activity of chrysin from oroxylum indicum via non-genotoxic p53 activation through the ATM-Chk2 pathway. Molecules. 23(1394)2018.PubMed/NCBI View Article : Google Scholar | |
|
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al: In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 303:844–848. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Schmitt CA, Wang B and Demaria M: Senescence and cancer-role and therapeutic opportunities. Nat Rev Clin Oncol. 19:619–636. 2022.PubMed/NCBI View Article : Google Scholar | |
|
Kong P, Yang X, Zhang Y, Dong H, Liu X, Xu X, Zhang X, Shi Y, Hou M and Song B: Palbociclib enhances migration and invasion of cancer cells via senescence-associated secretory phenotype-related CCL5 in non-small-cell lung cancer. J Oncol. 2022(2260625)2022.PubMed/NCBI View Article : Google Scholar | |
|
Nguyen HQ, To NH, Zadigue P, Kerbrat S, De La Taille A, Le Gouvello S and Belkacemi Y: Ionizing radiation-induced cellular senescence promotes tissue fibrosis after radiotherapy. A review. Crit Rev Oncol Hematol. 129:13–26. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Lee JY, Reyes NS, Ravishankar S, Zhou M, Krasilnikov M, Ringler C, Pohan G, Wilson C, Ang KKH, Wolters PJ, et al: An in vivo screening platform identifies senolytic compounds that target p16INK4a+ fibroblasts in lung fibrosis. J Clin Invest. 134(e173371)2024.PubMed/NCBI View Article : Google Scholar | |
|
Zannini L, Delia D and Buscemi G: CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol. 6:442–457. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Arienti KL, Brunmark A, Axe FU, McClure K, Lee A, Blevitt J, Neff DK, Huang L, Crawford S, Pandit CR, et al: Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J Med Chem. 48:1873–1885. 2005.PubMed/NCBI View Article : Google Scholar | |
|
Strzeszewska A, Alster O, Mosieniak G, Ciolko A and Sikora E: Insight into the role of PIKK family members and NF-кB in DNAdamage-induced senescence and senescence-associated secretory phenotype of colon cancer cells. Cell Death Dis. 9(44)2018.PubMed/NCBI View Article : Google Scholar | |
|
Zhao J, Zhang L, Lu A, Han Y, Colangelo D, Bukata C, Scibetta A, Yousefzadeh MJ, Li X, Gurkar AU, et al: ATM is a key driver of NF-κB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging (Albany NY). 12:4688–4710. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J and Elledge SJ: The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 349(aaa5612)2015.PubMed/NCBI View Article : Google Scholar | |
|
Nayak D, Kumar A, Chakraborty S, Rasool RU, Amin H, Katoch A, Gopinath V, Mahajan V, Zilla MK, Rah B, et al: Inhibition of Twist1-mediated invasion by Chk2 promotes premature senescence in p53-defective cancer cells. Cell Death Differ. 24:1275–1287. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Gire V, Roux P, Wynford-Thomas D, Brondello JM and Dulic V: DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 23:2554–2563. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Nakao M, Tanaka H and Koga T: Cellular senescence variation by metabolic and epigenomic remodeling. Trends Cell Biol. 30:919–922. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Bartek J and Lukas J: DNA damage checkpoints: From initiation to recovery or adaptation. Curr Opin Cell Biol. 19:238–245. 2007.PubMed/NCBI View Article : Google Scholar | |
|
Stracker TH, Usui T and Petrini JHJ: Taking the time to make important decisions: The checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst). 8:1047–1054. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Kandhaya-Pillai R, Miro-Mur F, Alijotas-Reig J, Tchkonia T, Schwartz S, Kirkland JL and Oshima J: Key elements of cellular senescence involve transcriptional repression of mitotic and DNA repair genes through the p53-p16/RB-E2F-DREAM complex. Aging (Albany NY). 15:4012–4034. 2023.PubMed/NCBI View Article : Google Scholar | |
|
Ha L, Ceryak S and Patierno SR: Chromium (VI) activates ataxia telangiectasia mutated (ATM) protein. Requirement of ATM for both apoptosis and recovery from terminal growth arrest. J Biol Chem. 278:17885–17894. 2003.PubMed/NCBI View Article : Google Scholar | |
|
Huart C, Fransolet M, Demazy C, Le Calvé B, Lucas S, Michiels C and Wéra AC: Taking advantage of the senescence-promoting effect of olaparib after X-ray and proton irradiation using the senolytic drug, ABT-263. Cancers (Basel). 14(1460)2022.PubMed/NCBI View Article : Google Scholar | |
|
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B: Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 282:1497–1501. 1998.PubMed/NCBI View Article : Google Scholar | |
|
Kastan MB and Bartek J: Cell-cycle checkpoints and cancer. Nature. 432:316–323. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Fennell DA, Porter C, Lester J, Danson S, Blackhall F, Nicolson M, Nixon L, Gardner G, White A, Griffiths G and Casbard A: Olaparib maintenance versus placebo monotherapy in patients with advanced non-small cell lung cancer (PIN): A multicentre, randomised, controlled, phase 2 trial. EClinicalMedicine. 52(101595)2022.PubMed/NCBI View Article : Google Scholar | |
|
Woll P, Gaunt P, Danson S, Steele N, Ahmed S, Mulatero C, Shah R, Bhosle J, Hodgkinson E, Watkins B and Billingham L: Olaparib as maintenance treatment in patients with chemosensitive small cell lung cancer (STOMP): A randomised, double-blind, placebo-controlled phase II trial. Lung Cancer. 171:26–33. 2022.PubMed/NCBI View Article : Google Scholar | |
|
Senra JM, Telfer BA, Cherry KE, McCrudden CM, Hirst DG, O'Connor MJ, Wedge SR and Stratford IJ: Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol Cancer Ther. 10:1949–1958. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Jiang Y, Verbiest T, Devery AM, Bokobza SM, Weber AM, Leszczynska KB, Hammond EM and Ryan AJ: Hypoxia potentiates the radiation-sensitizing effect of olaparib in human non-small cell lung cancer xenografts by contextual synthetic lethality. Int J Radiat Oncol Biol Phys. 95:772–781. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Coppé JP, Desprez PY, Krtolica A and Campisi J: The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol. 5:99–118. 2010.PubMed/NCBI View Article : Google Scholar | |
|
Soto-Gamez A and Demaria M: Therapeutic interventions for aging: The case of cellular senescence. Drug Discov Today. 22:786–795. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Hsu CH, Altschuler SJ and Wu LF: Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell. 178:361–373.e12. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Manila NG, Kaida A and Miura M: Kinetic analysis of radiation-induced cell-cycle alterations in HeLa cells expressing fluorescent ubiquitination-based cell cycle indicator (Fucci). Radiat Environ Med. 5:16–21. 2016. | |
|
Humar B, Müller H and Scott RJ: Cell cycle dependent DNA break increase in ataxia telangiectasia lymphoblasts after radiation exposure. Mol Pathol. 54:347–350. 2001.PubMed/NCBI View Article : Google Scholar |










