



TNF‑α induces premature senescence in tendon stem cells via the NF‑κB and p53/p21/cyclin E/CDK2 signaling pathways
- Authors:
- Published online on: July 10, 2025 https://doi.org/10.3892/ijmm.2025.5581
- Article Number: 140
-
Copyright: © Guo et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Achilles tendinitis (AT) commonly arises from trauma, iatrogenic factors and inflammatory conditions, particularly spondyloarthritis (1). Tendon ruptures have been reported in patients with systemic lupus erythematosus, often linked to corticosteroid therapy (2). In rheumatoid arthritis, tendinitis may involve the tendon, the surrounding synovium or both. Tendon involvement occurs in up to 64% of patients with rheumatoid arthritis, although some studies report lower prevalence rates (3,4). Typical symptoms of tendinitis include localized pain and tenderness along the affected tendon, particularly near joints, and symptoms often worsen with exercise or physical exertion (5). In AT, patients experience persistent pain and swelling accompanied by a sustained inflammatory response. Several inflammatory mediators contribute to the disease pathology, including tumor necrosis factor (TNF)-α, IL-1β, MMP and metabolic enzymes such as cyclooxygenase-2 (6-10).
Serum TNF-α levels are significantly higher in patients with rotator cuff tears accompanied by sleep disturbance compared with those with rotator cuff tears and normal sleep, as well as patients in a control group with chronic shoulder instability (11). The expression level of TNF-α is frequently upregulated in the subscapular sac tissue and degenerated tendon tissue of patients with rotator cuff injury (12,13). The overexpression of TNF-α in tendon sheath injuries can exacerbate the inflammatory response, thereby leading to more severe tissue damage and increased pain (12,13). Etanercept is a soluble fusion protein composed of two human tumor necrosis factor receptor II (TNFR II, molecular weight 75 kDa) subunits linked to the Fc portion of human IgG1. It is administered via subcutaneous injection and displays slow systemic absorption, reaching peak concentration 2-3 days post-injection (14). Acting as a decoy receptor, etanercept binds TNF-α and TNF-β with greater affinity than endogenous soluble TNFRs, thereby blocking their interaction with native receptors and preventing downstream proinflammatory signaling (15). By competitively inhibiting TNF-α binding to its natural receptors, etanercept disrupts inflammatory, apoptotic and immune-associated signaling pathways. This blockade effectively reduces inflammation in a range of pathological conditions (15-17). Experimental results demonstrated that etanercept treatment alleviated stress-shielding-induced structural and morphological changes in tendon collagen tissue (18). Etanercept suppressed the upregulation of MMP-13 and MMP-3, as well as collagen III expression levels (18).
Most adult human organs contain regenerative stem cells (SCs) that reside within specialized microenvironments, or niches, which are key for maintaining tissue homeostasis and facilitating repair following injury (19). The function of SCs depends primarily on their dynamic interaction with the surrounding microenvironment. Tendon SCs (TSCs) are undifferentiated cells within tendon tissue, characterized by their ability to self-renew and differentiate into tenocytes (20). Although studies suggest that TSCs contribute to tendon repair and regeneration during disease progression, the underlying molecular mechanisms remain poorly understood (20-22).
TNF-α is a key cytokine involved in inflammation. TNF-α not only activates proinflammatory genes but also exacerbates inflammatory responses by promoting apoptosis, modulating immune activity and facilitating disease progression (23). Elevated TNF-α levels are commonly observed in inflammatory diseases, where they influence the development, senescence and functional properties of mesenchymal SCs (MSCs) under specific physiological and pathological conditions (24). However, the role of TNF-α in the onset and progression of tendon disorders remains insufficiently defined. While anti-TNF-α biological agents have proven highly effective in treating chronic inflammation and autoimmune disease, their effects on TSCs are still unclear (25). Therefore, investigating the influence of TNF-α and TNF-α inhibitors on TSCs is key.
The present study aimed to clarify the role of TNF-α in tendon pathology by examining its effects on TSC senescence and inflammation at cellular and animal levels, and to assess whether TNF-α inhibition with Etanercept could mitigate these pathological processes. This work seeks to provide mechanistic insights into tendon disorders and validate potential therapeutic strategies.
Materials and methods
Cell isolation and culture
All animal procedures strictly followed institutional regulations for animal research and were approved by the Animal Care Committee of Nantong University (Nantong, China; approval no. S20230522-005). TSCs were isolated from the flexor digitorum profundus (FDP) tendons of female C57BL/6 mice (n=6, weight 16-18 g, provided by Nantong University Experimental Animal Center, Nantong, China; age, 4 weeks). The mice were euthanized humanely via cervical dislocation. FDP tendons were dissected from the index, middle and ring digits of the hind limbs, washed in sterile phosphate-buffered saline (PBS) and finely minced into small fragments. Tissue was digested using a solution containing 3 mg/ml type I collagenase and 4 mg/ml dispase (Gibco, Thermo Fisher Scientific, Inc.) for 2 h at 37°C. The cell suspension was filtered through a 70-µm cell strainer to obtain a single-cell suspension, which was seeded onto 25-cm2 culture plates. Cells were cultured in DMEM supplemented with 10% FBS (both Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin. Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. Medium was replaced every 3 days to support optimal cell proliferation. Cells at 85-90% confluence were sub-cultured at a ratio of 1:3 (one part cells was split into three new culture vessels). Cells were detached using 0.25% trypsin and transferred to new flasks for continued expansion and downstream analyses.
Cells were grouped as follows (all treatments were performed at 37°C): i) TSCs treated with PBS served as the control; TSCs stimulated with TNF-α at ii) 20 and iii) 40 ng/ml for 6 h, repeated three times at 48-h intervals and TSCs were stimulated with TNF-α at iv) 20 and v) 40 ng/ml for 6 h, repeated six times at 24-h intervals. To investigate the potential therapeutic effect of TNF-α inhibition, TSCs stimulated three times with 20 ng/ml TNF-α-were co-cultured with a TNF-α antagonist (Etanercept) for 48 h at 37°C. For the therapeutic co-culture assay, TSCs pre-stimulated three times with 20 ng/ml TNF-α (37°C, 6 h per stimulation, 48-h intervals) were treated with the TNF-α antagonist etanercept (recombinant human TNF-α receptor II:IgG Fc fusion protein, 20 ng/ml, supplied by Sunshine Guojian Pharmaceutical) at 37°C for 48 h. Untreated TSCs served as controls.
Differentiation assay
TSCs were seeded at a density of 1×104 cells/cm2 to initiate differentiation. Mouse TSCs were induced to undergo osteogenic and adipogenic differentiation using a commercial differentiation medium kit (cat. no. RASTA-90021; cat. no. RASTA-90031, Cyagen Biosciences, Inc.). Cells were cultured in differentiation media at 37°C with 5% CO2 for 28 days. Following washing with PBS, cells were fixed in 4% PFA) at room temperature for 15 min. To assess osteogenic differentiation, cells were stained with Alizarin Red S and underwent an alkaline phosphatase assay (cat. no. C0148S, cat. no. C3250S, Beyotime Institute of Biotechnology) at room temperature for 20 min to evaluate matrix calcification. 0.5% Oil Red O staining (cat. no. C0157M, Beyotime Institute of Biotechnology) at room temperature for 20 min was performed to detect intracellular neutral lipid vacuoles, indicating adipogenic differentiation.
To initiate chondrogenic differentiation, TSCs were centrifuged in 15 ml conical tubes at 160 × g at room temperature for 5 min to form cell pellets. Pellets were cultured in chondrogenic differentiation medium (cat. no. RASTA-90041, Cyagen Biosciences, Inc.) at 37°C with 5% CO2, replacing the medium every 3 days for a total of 4 weeks. Pellets were fixed in 4% PFA at room temperature for 24 h, dehydrated through a graded sucrose series 4°C, 4 h per step, embedded in optimal cutting temperature compound, and sectioned into 5-µm slices. Sections were stained with toluidine blue (cat. no. C0637, Beyotime Institute of Biotechnology) at room temperature for 10 min for histological evaluation. Images were captured using a light microscope (Leica).
Senescence associated β-galactosidase (SA-β-gal) assay
TSC senescence was assessed using the Senescence β-Galactosidase Staining kit (SA-β-gal assay kit) (cat. no. C0602, Beyotime Institute of Biotechnology), according to the manufacturer's instructions. TSCs were cultured at a density of 1×106 cells/well in 6-well plates and fixed with 4% PFA at room temperature for 10 min following treatment as aforementioned. Cells were incubated at 37°C overnight with the SA-β-gal staining solution. Following thorough washing by PBS, cells were examined under a light microscope (Olympus) at ×10 magnification. Senescent cells exhibited blue staining. To quantify senescence, 1,000 cells were counted across 20 randomly selected fields of view/slide and the percentage of SA-β-gal-positive cells was calculated (26).
Immunofluorescent staining
TSCs were fixed in 4% PFA at 4°C for 1 h, then washed with 0.1% Triton X-100 in PBS (PBST). Cells were blocked in PBST containing 10% FBS at room temperature for 30 min and incubated overnight at 4°C with primary antibodies (1:100) targeting γ-H2A.X (cat. no. ab81299, anti-rabbit, Abcam), p65 (cat. no. sc-8008, anti-mouse, Santa Cruz Biotechnology, Inc.), p53 (cat. no. ab246550, anti-rabbit, Abcam), p21 (cat. no. ab188224, anti-rabbit, Abcam), Oct4 (cat. no. ab200834, anti-rabbit, Abcam) and Nanog (cat. no. ab109250, anti-rabbit, Abcam). Following washing with PBS, secondary antibodies [goat Anti-Mouse IgG H&L (Alexa Fluor® 488) (ab150113), Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488) (ab150077), goat Anti-Rabbit IgG H&L (Alexa Fluor® 568) (ab175471), Abcam] at 1:250 were added for 2 h at room temperature. Nuclei were counterstained with DAPI (cat. no. ab228549, Abcam) at 1:500 at room temperature for 30 min and cells were examined using a fluorescence microscope at ×20 magnification.
Filamentous (F-)actin staining
The Cell Navigator™ F-Actin Labeling kit (cat. no. 22661, AAT Bioquest) was used to visualize F-actin) in fixed cells, according to the manufacturer's instructions. TSCs were fixed in 4% PFA at 4°C for 30 min, followed by washing with PBS at room temperature. Cells were permeabilized with 0.1% PBST at room temperature for 5 min and incubated with iFluor™ 488-Phalloidin working solution at room temperature for 30 min. After washing with PBS, nuclei were counterstained with DAPI (cat. no. ab228549, Abcam) at 1:500 at room temperature for 30 min and cells were examined using a fluorescence microscope at ×20 magnification.
Reactive oxygen species (ROS) staining
ROS staining kit (cat. no. S0033S, Beyotime Institute of Biotechnology) was used according to the manufacturer's instructions. After washing three times with PBS, TSCs were incubated with ROS-sensitive dye DCFH-DA at room temperature for 30 min. Following another PBS wash, nuclei were counterstained with DAPI (cat. no. ab228549, Abcam) at 1:500 at room temperature for 30 min and cells were examined using a fluorescence microscope at ×20 magnification.
EdU cell proliferation assay
TSC proliferation was assessed using the Cell-Light™ EdU Apollo In Vitro kit (Guangzhou RiboBio Co., Ltd.), following the manufacturer's protocol. TSCs in the logarithmic growth phase were seeded into 24-well plates at a density of 2×104 cells/well and cultured to 70% confluency). Then, 25 µM EdU medium was added to each well at 37°C for 24 h. Cells were washed twice with PBS and fixed with 4% PFA at room temperature for 30 min, treated with 2 mg/l glycine at room temperature for 5 min, washed twice with PBS for 5 min and permeabilized with Triton X-100 (cat. no. P0096, Beyotime Institute of Biotechnology) for 10 min at room temperature. Apollo® reaction mixture was added at room temperature for 30 min and cells were washed twice with PBS at room temperature. Finally, nuclei were stained with DAPI (cat. no. ab228549, Abcam) at 1:500 at room temperature for 10 min before washing with PBS and imaging the cells by fluorescence microscopy at ×10 magnification.
Cell cycle detection
Cell cycle distribution was assessed using the Cell Cycle Assay kit (Beijing Solarbio Science & Technology Co., Ltd.) according to the manufacturer's instructions. Cells were fixed in pre-chilled 95% ethanol at 4°C for 24 h, washed with PBS and stained with propidium iodide at 37°C for 30 min. DNA content was analyzed by flow cytometry using a FACSCalibur system (BD Biosciences) and cell cycle distribution was quantified using ModFit LT software (version 5.0, Verity Software House, vsh.com/).
Western blotting
TSCs were lysed for 30 min using RIPA buffer containing protease inhibitors (cat. no. P0013B, Beyotime Institute of Biotechnology). Supernatant was collected following centrifugation at 12,800 × g 4°C for 20 min. Protein concentration was quantified use BCA assay, samples were mixed with loading buffer and protein was denatured by heating at 100°C for 10 min. Protein (20 µg/lane) was separated by 10% or 12% SDS-PAGE and transferred onto PVDF membranes using a BIO-RAD transfer system at 300 mA. Membranes were blocked with 5% skimmed milk at room temperature for 2 h, then incubated overnight at 4°C with primary antibodies (1:1,000). After washing by Tris-Buffered Saline with Tween 20 (TBST), membranes were incubated with secondary antibodies (1:10,000) (cat. no. D110087, D110058, Sangon Biotech) at room temperature for 2 h. Primary antibodies included GAPDH (cat. no. 10494-1-AP, anti-rabbit, Proteintech Group, Inc.), p65 (cat. no. sc-8008, anti-mouse, Santa Cruz Biotechnology), p53 (cat. no. ab26, anti-mouse, Abcam), p21 (cat. no. ab188224, anti-rabbit, Abcam), phosphorylated p65 (p-p65; cat. no. sc-136548, anti-mouse, Santa Cruz Biotechnology), γ-H2A.X (cat. no. ab81299, anti-rabbit, Abcam), CDK2 (cat. no. ab32147, anti-rabbit, Abcam) and cyclin E (cat. no. 11554-1-AP, anti-rabbit, Proteintech Group, Inc.), H2A.X (cat. no. 10856-1-AP, anti-rabbit, Proteintech Group, Inc.). The membranes were incubated with anti-IgG secondary antibodies conjugated to horseradish peroxidase (HRP). Detection of specific bands was performed using an ECL Detection kit (Bio-Rad), and visualization was carried out with the ChemiDoc XRS + System (Bio-Rad). The densitometry was performed using ImageJ software (version 1.53t, National Institutes of Health).
Tendinopathy rat model
Rats were housed in a specific pathogen-free (SPF) facility at temperature 20-26°C, humidity 30-70%, with a 12-h light/dark cycle and provided ad libitum access to standard rodent chow and tap water. A total of 30 male Sprague-Dawley rats (age, 8 weeks; weight, 220±9 g, Nantong University Experimental Animal Center, Nantong, China) were randomly assigned to three treatment groups (n=10/group), each receiving injections in the Achilles tendon: i) PBS control; ii) PBS + collagenase type I (5 mg/ml) and iii) collagenase type I (5 mg/ml)+ etanercept (0.5 mg/kg). At 20 days after collagenase type I (5 mg/ml) treatment twice, administered 7 days apart, etanercept (0.5 mg/kg) was administered seven times over 2 weeks. Achilles tendons were harvested for histopathological analysis to evaluate collagenase-induced tendinopathy severity and the therapeutic effects of etanercept (27). The dose of etanercept (0.5 mg/kg) was selected based on Chen et al (18), who investigated that in a rat model of stress-shielded tendons, etanercept at 0.5 mg/kg/week for 2 weeks significantly attenuated early tendon degeneration by reducing collagen fibril disorganization and suppressing matrix metalloproteinase (MMP)-13, MMP-3, and collagen III expression. This dosing strategy was adopted to target tumor necrosis factor-α (TNF-α)-mediated inflammation, the key pathway identified in both stress-shielding and collagenase-induced tendinopathy models (18).
Rats were anesthetized with isoflurane (3.0-3.5% in oxygen, 500-700 ml/min) until completely unresponsive to paw pinch. Following anesthesia, death was induced by CO2 asphyxia (40% vol/min for 5 min) followed by cervical dislocation, consistent with the AVMA Guidelines for the Euthanasia of Animals (28). Death was verified by the absence of corneal reflex, breathing and heartbeat.
Histological analyses
Tendon specimens were fixed in 4% PFA at 4°C for 24 h. Samples were dehydrated through graded ethanol, embedded in paraffin and cut into longitudinal sections of 4.5 µm thickness. Sections were stained at room temperature with hematoxylin and eosin (H&E) and Masson's Trichrome to assess tendon morphology. For H&E staining, tissue sections were dewaxed and rehydrated. Nuclei were stained with hematoxylin for 5 min. Differentiation was performed in acid alcohol for 30 sec. Cytoplasmic counterstaining was achieved using eosin for 1 min, and sections were rapidly dehydrated through ascending ethanol gradients. Sections were assed using a light microscope ×20 and ×40 magnification. For Masson's Trichrome staining, the sections were mordanted by immersion in mordant solution at 60°C for 1 h. Sections were then stained with celestin blue solution for 3 min, stained with Mayer's hematoxylin for 3 min. Differentiation was performed in acid solution, followed by a 10-min tap water rinse for bluing. Sections were stained with Ponceau-fuchsin for 10 min. Phosphomolybdic acid treatment for 10 min was followed by direct application of aniline blue solution for 5 min without intermediate washing. Excess aniline blue was removed with weak acid solution, and sections were further treated with weak acid for 2 min. Rapid dehydration was performed in ethanol. Sections were assed using a light microscope ×10 and ×20 magnification.
For immunohistochemistry, paraffin-embedded sections were treated with primary antibodies against TNF-α (1:200, ab183218, Abcam) at 4°C overnight, then incubated with secondary antibodies enzyme-labeled goat anti-mouse/rabbit IgG polymer (PV-6000, ZSGB-BIO) for 20 min at room temperature. Tendon specimens were rapidly frozen in liquid nitrogen and sectioned into 4.5 µm slices. Thawed sections were washed three times with PBS and fixed in β-galactosidase staining fixing solution (cat. no. R0757, Shifeng Biological) at room temperature overnight. After rinsing with PBS, β-Gal Staining A Solution 1 µl, β-Gal Staining B Solution 1 µl, β-Gal Staining C Solution 93 µl and X-Gal Solution 5 µl) was added for 24 h at 37°C. Sections were assed using a light microscope (29).
For immunofluorescence staining, tendon samples were incubated with 0.1% PBS-Triton X at room temperature for 15 min to permeabilize the tissue, followed by blocking (cat. no. P0260, Beyotime Institute of Biotechnology, Shanghai, China) to prevent non-specific antibody binding at room temperature for 20 min. Sections were incubated overnight at 4°C with primary antibodies against CD44 (cat. no. 15675-1-AP, Proteintech, 1:100) and p53 (cat. no. 60283-2-Ig, Proteintech, 1:100). The samples were incubated with secondary antibodies [Goat Anti-Mouse IgG H&L (Alexa Fluor® 568) (ab175473), Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488) (ab150077), Abcam] at 1:250 for 2 h, followed by nuclear staining with DAPI (cat. no. ab228549, Abcam) at 1:500 at room temperature. Sections were analyzed using a fluorescence microscope (BX41, Olympus Corporation).
Statistical analysis
All data are presented as the mean ± SEM from ≥3 independent experiments, each performed in triplicate. Statistical analyses were conducted using SPSS 16.0 (SPSS, Inc.). For comparisons involving three or more groups, data were evaluated by one-way ANOVA followed by Tukey's post hoc test for pairwise comparisons. For two-group comparisons, Student's unpaired t-test was used, as all samples were derived from independent experimental units. P<0.05 was considered to indicate a statistically significant difference.
Results
TSCs exhibit SC-like properties and TNF-α induces senescence in TSCs
TSCs displayed a spindle-shaped, fibroblast-like morphology and multipotent differentiation capacity toward osteogenic, adipogenic and chondrogenic lineages (Fig. 1A-D). The cells also showed positive expression of stemness markers Oct4 and Nanog (Fig. 1E). TNF-α intensified SA-β-gal staining, caused progressive cell enlargement and reduced the typical spindle-shaped morphology. Following treatment with TNF-α, TSCs exhibited an expanded and flattened phenotype (Fig. 2A). The proportion of SA-β-gal-positive TSCs increased with higher TNF-α concentrations and longer stimulation durations (Fig. 2A and B). Immunofluorescence revealed disrupted F-actin organization and elevated expression (Fig. 2C). EdU staining showed a significant decrease in proliferative capacity in TSCs treated with TNF-α compared with controls (Fig. 2D and E). Since cell cycle arrest is a hallmark of cellular senescence (30), flow cytometry was performed on TNF-α-stimulated TSCs, which revealed an increased proportion of cells in the G0/G1 phase (Fig. 2F and G). Collectively, these findings indicate that TSC senescence increased with higher TNF-α concentration and stimulation duration.

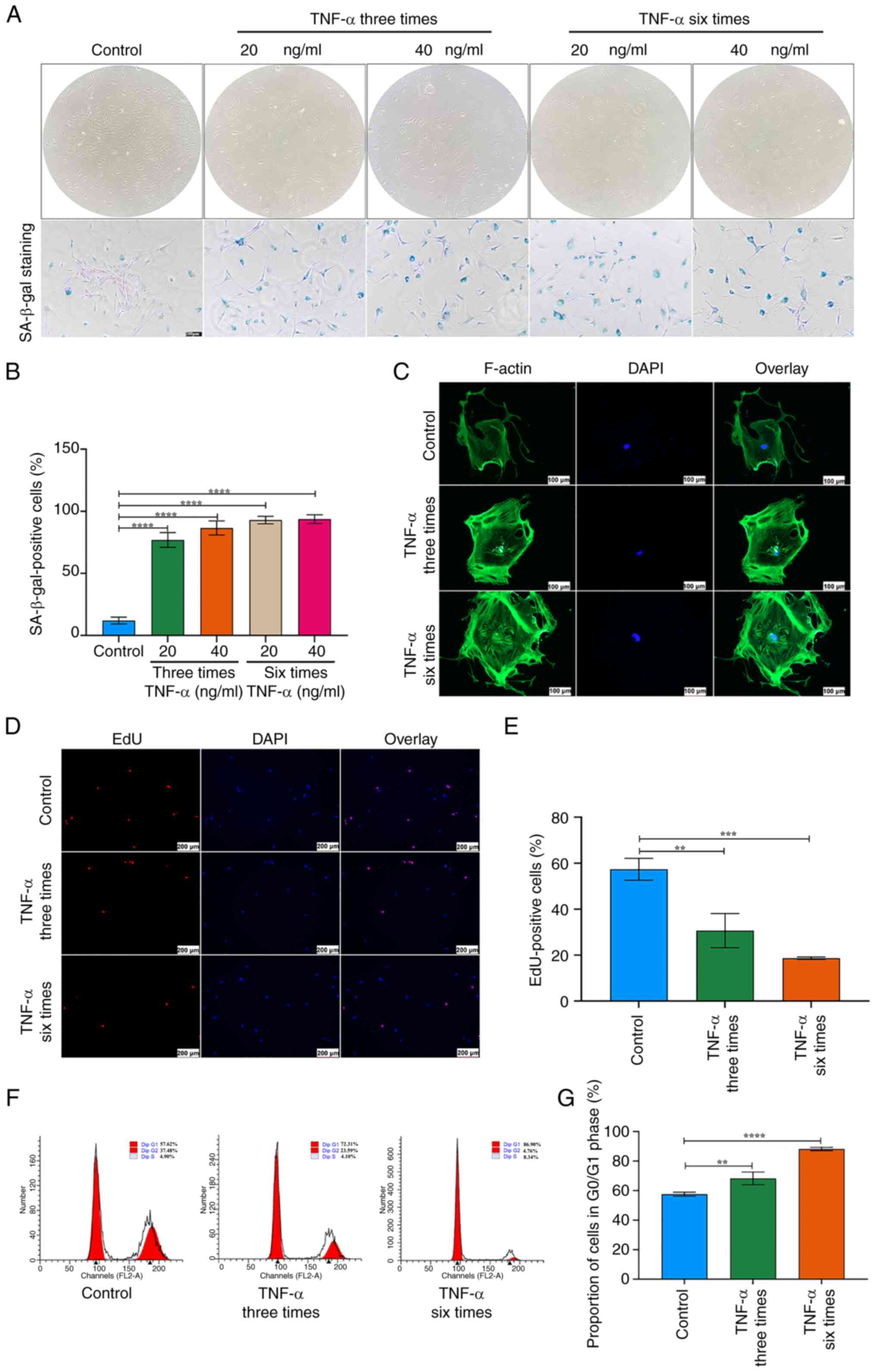
TNF-α promotes ROS production and DNA damage in TSCs
As ROS are inducers of cellular senescence (31), intracellular ROS levels were measured in TSCs treated with TNF-α. TNF-α significantly elevated ROS production in TSCs (Fig. 3A and B). To evaluate the effect of increased ROS on the DNA damage response, expression of γ-H2A.X, a specific marker of DNA damage foci, was assessed. Immunofluorescence showed that γ-H2A.X levels increased following TNF-α stimulation (Fig. 3C and D). The γ-H2A.X expression increased after TNF-α stimulation, indicating enhanced DNA damage. H2A.X expression did not differ significantly between the two groups, as confirmed by western blot analysis (Fig. 3E and F). These results suggested that TNF-α treatment enhances ROS generation, which is associated with increased DNA damage in TSCs.

TNF-α promotes expression of NF-κB and p53/p21/cyclin E/CDK2 signaling pathway components during senescence in TSCs
Following TNF-α stimulation, TSCs exhibited increased expression of p-p65, while total p65 proteins levels showed no significant difference (Fig. 3E and F). The ratio of p-p65 to total p65 demonstrated a significant upregulation (Fig. 3E and F). Compared with the control group, TNF-α at concentrations of 20 and 40 ng/ml, elevated the expression of p53 and p21. TNF-α (20 ng/ml, three or six times) all elevated the expression of p53; however, these changes did not follow frequency-dependent pattern. TNF-α at 20 ng/ml, administered either three or six times, significantly elevated p21 expression. Notably, six administrations induced a higher p21 expression than three administrations (Fig. 3G and H). TNF-α suppressed the expression of cyclin E and CDK2 in TSCs (Fig. 3G and H). Immunofluorescence staining demonstrated nuclear translocation of p65 following TNF-α exposure (Fig. 3I). Additionally, immunofluorescence revealed a marked increase in the number of p53- and p21-positive cells in TNF-α-treated TSCs (Fig. 3I). These results indicated that TNF-α promoted TSC senescence by modulating NF-κB signaling and altering the p53/p21/cyclin E/CDK2 regulatory pathway.
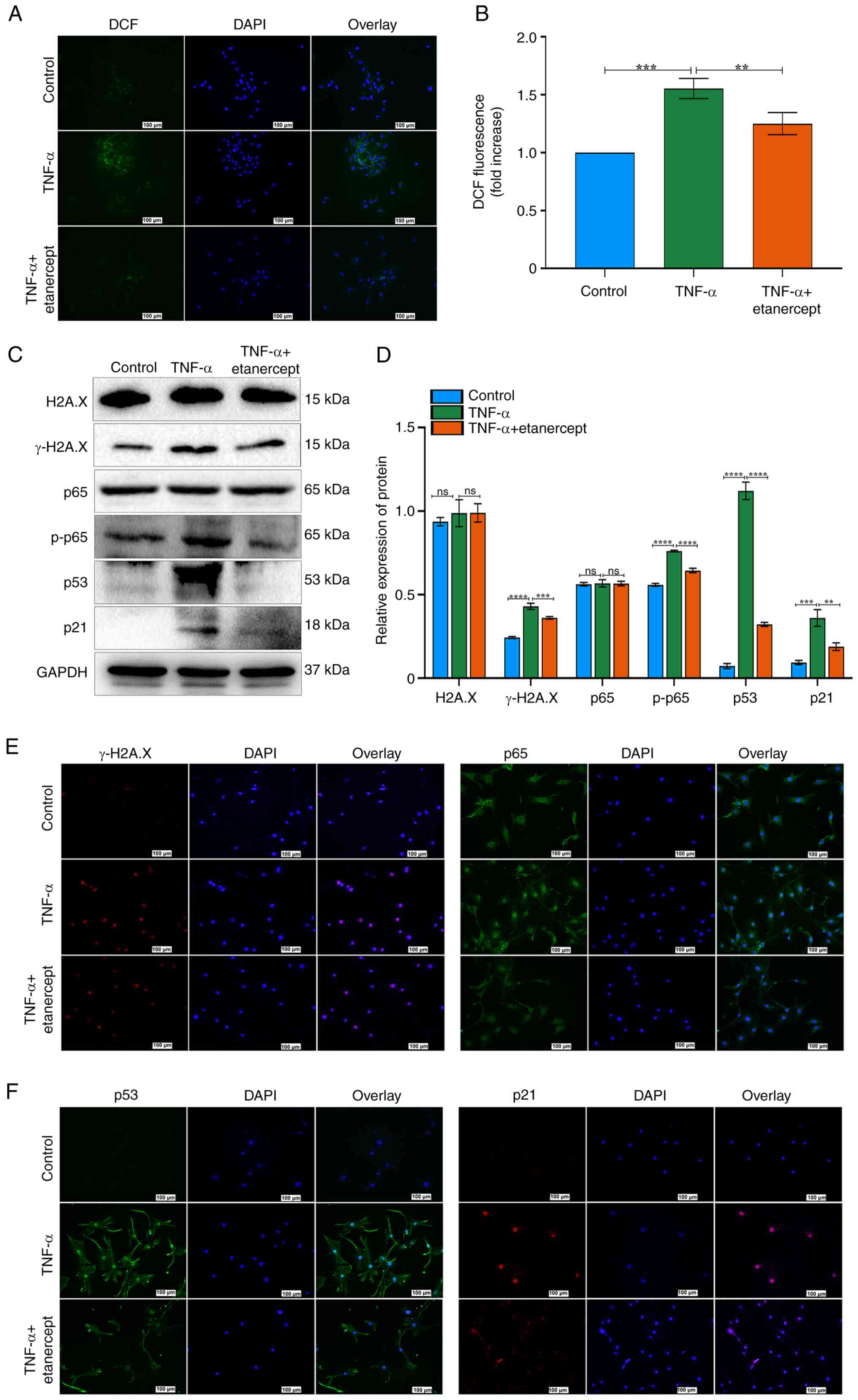
Effects of etanercept on the NF-κB and p53/p21/cyclin E/CDK2 signaling pathways in TNF-α-Induced TSCs Senescence
To investigate the role of the NF-κB and p53/p21/cyclin E/CDK2 signaling in TNF-α–induced senescence, TSCs were treated with the TNF-α antagonist etanercept. TNF-α increased ROS fluorescence, while etanercept significantly reduced this effect (Fig. 4A and B). These results indicated that etanercept possesses antioxidant activity capable of attenuating TNF-α-induced oxidative stress. Etanercept also decreased γ-H2A.X expression and DNA damage in TSCs (Fig. 4C-E). H2A.X expression did not differ significantly between the two groups, as confirmed by Western blot analysis (Fig. 4C and D). Western blot analysis confirmed that etanercept reversed TNF-α-induced senescence, as evidenced by decreased expression of p53, p21 and p-p65 (Fig. 4C and D). By contrast, total p65 levels remained unchanged (Fig. 4C and D). Etanercept inhibited the nuclear translocation of p65 in response to TNF-α (Fig. 4E). Immunofluorescence staining also revealed decreased nuclear accumulation of p53 and p21 in the TNF-α + etanercept group (Fig. 4F). Etanercept, a TNF inhibitor (recombinant Human TNF-α Receptor II: IgG Fc Fusion Protein) effectively reversed TNF-α-induced senescence in TSCs. Together, these findings suggest that etanercept attenuates TNF-α-induced senescence in TSCs by modulating oxidative stress and inhibiting key components of the NF-κB and p53/p21 signaling pathways.
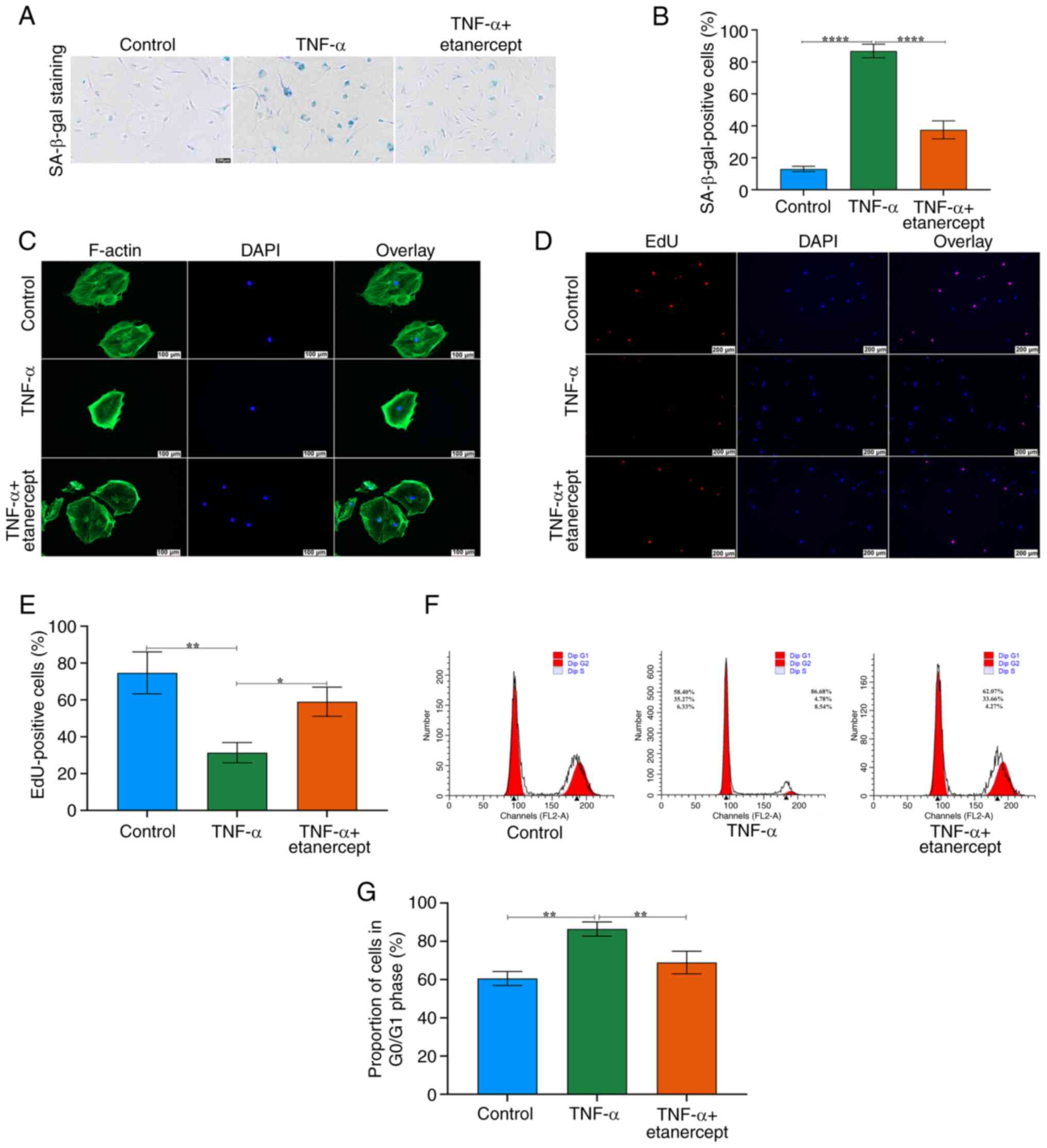
TNF-α antagonists reverse TNF-α-induced senescence in TSCs
To evaluate the therapeutic potential of TNF-α inhibition, TSCs pre-treated with TNF-α (20 ng/ml, three cycles) were co-cultured with Etanercept (20 ng/ml). Etanercept significantly decreased the number of SA-β-gal-positive cells (Fig. 5A and B). Etanercept markedly restored F-actin cytoskeleton integrity disrupted by TNF-α, normalizing F-actin morphology (Fig. 5C). EdU staining confirmed that etanercept rescued the proliferative capacity of TSCs (Fig. 5D and E). Etanercept normalized the ratio of cells in the G1/G0 phase, indicating restoration of normal cell cycle progression (Fig. 5F and G).
Histological changes in AT rats
To evaluate the therapeutic effects of local etanercept injection in AT, a rat model was established by administering collagenase I (Fig. 6A). H&E staining of tendons revealed notable inflammatory cell infiltration (Fig. 6B). To assess tendon repair, Masson's trichrome staining was performed 2 weeks after local injection with either PBS or etanercept (Fig. 6B). Tendons in the collagenase I + PBS group showed disrupted and disorganized collagen fiber architecture, while etanercept treatment mitigated collagen degradation and improved fiber alignment (Fig. 6B).

Immunohistochemistry revealed that collagenase I increased TNF-α protein levels, whereas etanercept significantly suppressed TNF-α expression (Fig. 6C and D). Moreover, the proportion of SA-β-gal-positive cells was significantly elevated in the collagenase I + PBS group compared with PBS controls, but etanercept treatment decreased levels of this senescence marker (Fig. 6E and F). Finally, double immunostaining demonstrated increased p53 expression in CD44-positive TSCs within tendon tissue, confirming cellular senescence in TSCs (Fig. 6G).
Discussion
Previous studies have established TNF-α as a key mediator in the pathogenesis of AT (18,32-34). Elevated TNF-α levels are consistently reported in tendon injury models and in the serum and tissue samples of patients with tendinopathy (18,32-34). Beyond its role in promoting inflammation, TNF-α is a target for regenerative therapies (18). Gulotta et al (35) demonstrated that polyethylene glycol-conjugated soluble TNF receptor I enhances collagen deposition and improves biomechanical properties of healing tendons in a rat rotator cuff repair model, high-lighting its potential to support tendon regeneration. Similarly, Chen et al (18) showed that etanercept decreases inflammation and promotes the proliferation of tendon-associated cells, contributing to improved tendon healing outcomes. Despite these findings, the specific effects of TNF-α on TSCs remain poorly defined. While previous studies have broadly characterized the role of TNF-α in tendon pathophysiology (36,37), they have not fully explored how TNF-α influences TSC stemness, proliferation or differentiation at the molecular level. The present study aimed to systematically investigate the cellular and molecular responses of TSCs to TNF-α exposure. The present study assessed TNF-α-induced cellular senescence, ROS accumulation, DNA damage and the activation of key signaling cascades, including the NF-κB and p53/p21/cyclin E/CDK2 pathways, and evaluated the therapeutic potential of etanercept in reversing TNF-α-induced senescence in TSCs. The present findings provide new mechanistic insight into the role of TNF-α in TSCs dysfunction and suggest a promising avenue for targeting AT through TNF-α inhibition.
TSCs exhibit a spindle-shaped, fibroblast-like morphology and possess the capacity to differentiate into cartilage, adipose tissue and bone (38). Emerging evidence suggests that pro-inflammatory cytokines promote cell senescence (39). For example, IL-6 induces senescence in telomerase-inhibited growth 3 (TIG3) fibroblasts by activating the STAT3/ IGF binding protein 5 (IGFBP5) signaling pathway, increasing ROS levels and promoting a senescent phenotype (40). Similarly, TNF-α drives senescence in proliferative endothelial progenitor cells via activation of the p38 MAPK pathway (41). Previous studies have demonstrated that TNF-α not only amplifies inflammation by upregulating inflammatory gene expression but also contributes to disease onset and progression by inducing apoptosis and activating immune responses in target cells (42,43). The present study used TNF-α concentrations of 20 and 40 ng/ml based on previous reports and preliminary experiments (44,45). Research involving MSCs exposed to inflammatory cytokines has shown that TNF-α concentrations within this range effectively induce inflammation-associated cell responses (46). For example, studies investigating the role of TNF-α in MSCs under inflammatory conditions used similar concentrations (10-40 ng/ml) to elicit significant changes in senescence-associated markers, such as p21 and SA-β-galactosidase activity (47,48). Here, increasing the concentration and frequency (3 vs. 6 times) of TNF-α stimulation led to a progressive rise in the number of senescent TSCs. This increase was associated with reduced stemness, enlarged cell morphology, intensified SA-β-gal staining, impaired proliferative capacity and an elevated proportion of cells in the G1/G0 phase (49-51). Moreover, TNF-α disrupted cell cycle progression in TSCs. Notably, treatment with a TNF-α inhibitor restored both the proliferative ability and SC characteristics of TSCs, highlighting the potential of TNF-α inhibition as a therapeutic strategy.
Any signaling pathway that drives inflammation should also influence inflammation-associated senescence. However, understanding of the molecular mechanisms underlying inflammatory senescence remains limited, underscoring the need for further investigation. NF-κB is a multifaceted nuclear transcription factor that is broadly expressed in various tissues and cell types (52). Upon activation, it regulates the expression of numerous genes involved in immune responses, inflammation, oxidative stress, cell proliferation and apoptosis (53). NF-κB also serves a key role in the intracellular signaling processes frequently activated in senescent SCs (54). The present in vitro experiments demonstrated that TNF-α activates NF-κB signaling in TSCs, as evidenced by increased p-p65 expression. However, the extent to which NF-κB mediates TSC senescence in vivo remains unclear. Wang et al (55) reported that inhibiting the IκB kinase-β) /NF-κB signaling axis reverses inflammaging (chronic low-grade inflammation associated with aging)-induced TSC senescence and promotes tendon healing in a rat model. Consistent with this, the present findings support a link between TNF-α stimulation and NF-κB activation in TSCs. Whether TNF-α drives TSC senescence exclusively through NF-κB signaling remains to be determined. Future studies should use a specific NF-κB inhibitor to assess the extent of its involvement in TNF-α-induced TSC senescence. Although prior studies have implicated NF-κB in tendon degeneration (56,57), the present data suggest that TNF-α exerts broader effects on the tendon microenvironment, including promotion of inflammation, disruption of extracellular matrix organization and impairment of SC function. Future studies should use genetic or pharmacological strategies to investigate the contributions of NF-κB and other downstream mediators in TNF-α-driven tendon pathology.
Comparing etanercept with other TNF-α inhibitors reveals differences in molecular structure, pharmacokinetics and side-effect profiles. The distinctive fusion-protein design of etanercept grants it a longer half-life, allowing for less frequent dosing than some alternatives, such as methotrexate (58,59). However, choosing a TNF-α inhibitor depends on factors such as disease type, patient characteristics and treatment response. Here, etanercept effectively reversed TNF-α-induced senescence in TSCs and improved tendon histology in a rat model. Future studies should focus on optimizing etanercept dosing for tendinitis treatment, assessing its long-term effects on tendon function and regeneration, and investigating potential synergistic effects when combined with growth factors or anti-inflammatory agents to enhance therapeutic outcomes. Translating these preclinical findings into clinical trials will be critical to establish the practical efficacy of etanercept in managing AT.
The present in vivo data offer novel insights into the pathological role of TNF-α in tendonitis and highlight the therapeutic potential of TNF-α inhibitors. In the rat AT model, histological analysis showed that etanercept reduced inflammatory cell infiltration, collagen fiber disarray and matrix degradation. These results corroborate previous studies demonstrating that TNF-α inhibitors effectively decrease tendon inflammation and enhance tissue repair (18,35). Importantly, the decrease in TNF-α expression following etanercept treatment confirmed direct modulation of the TNF-α pathway in tendon tissue. Clinically, TNF-α inhibitors may offer a promising therapeutic strategy for AT. While current treatments mainly target symptom relief, inhibiting TNF-α may address the underlying inflammatory processes that drive tendon degeneration. Nevertheless, challenges remain before clinical application. Determining optimal dosing, treatment duration and long-term safety require further study. Moreover, the diverse etiologies of tendinitis, including overuse and systemic inflammation, may demand personalized therapeutic approaches.
Future studies should investigate the long-term effects of TNF-α and etanercept on TSCs, with a focus on elucidating the crosstalk between key signaling pathways. Evaluating the therapeutic potential of etanercept in patients with AT is required to translate preclinical findings into clinical application. Additionally, combination therapies involving etanercept and other pharmacological or biological agents to enhance therapeutic efficacy should be investigated to identify the role of TNF-α in tendon pathology and support the development of more effective, targeted treatment strategies.
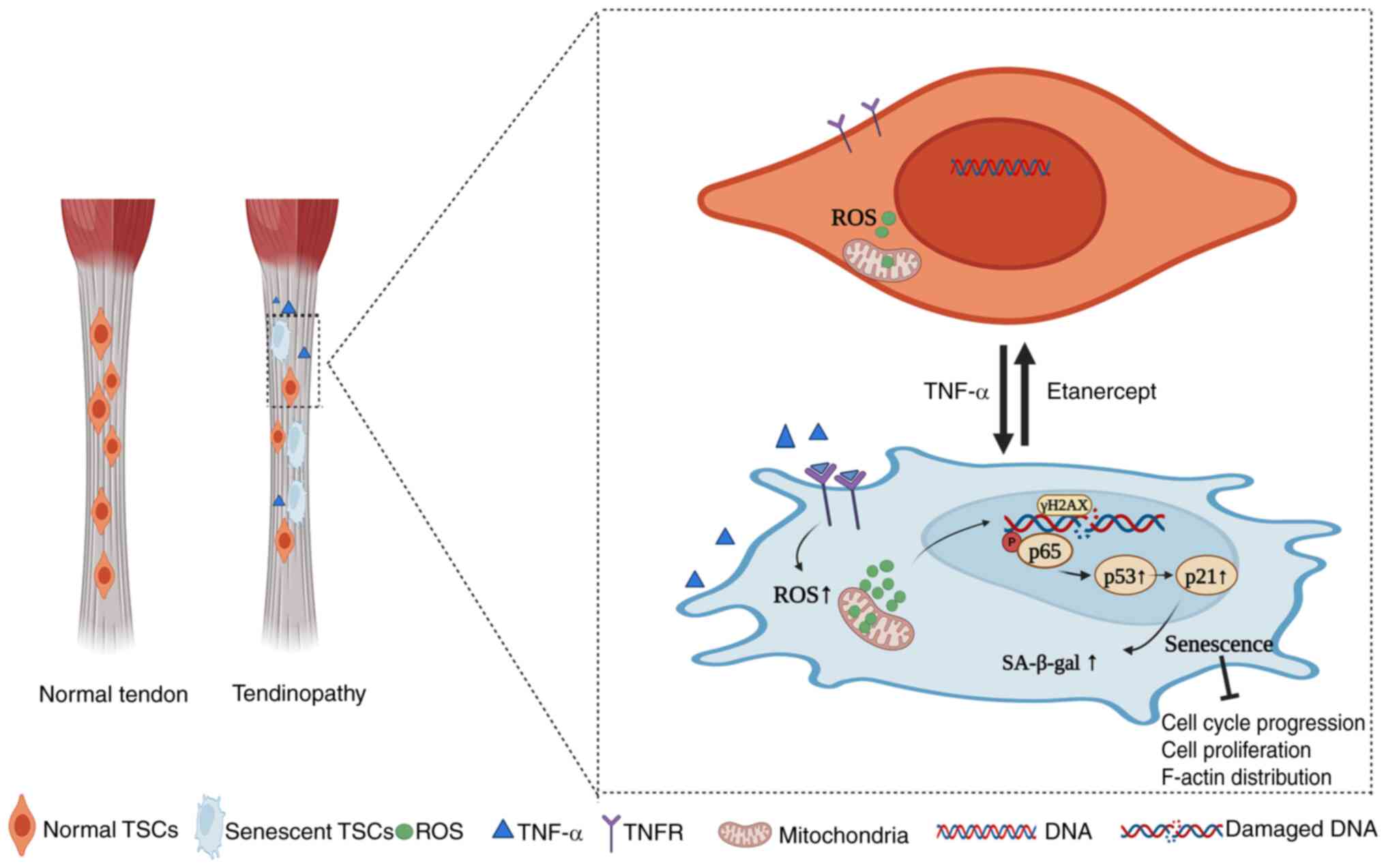
In conclusion, the present study demonstrated that TNF-α induced premature senescence in TSCs, as demonstrated by increased SA-β-gal activity, disrupted F-actin cytoskeleton and cell cycle arrest in the G0/G1 phase. Mechanistically, TNF-α activates the NF-κB signaling pathway, as evidenced by the significant upregulation of p-p65 and its nuclear translocation in TSCs. This activation is further linked to the induction of DNA damage (γ-H2A.X expression) and upregulation of the p53/p21/cyclin E/CDK2 pathway, which collectively drive cellular senescence. TNF-α antagonist etanercept reverses the senescent phenotype, as shown by reduced SA-β-gal-positive cells, restored F-actin cytoskeleton organization, and normalized cell cycle progression. These effects are linked to Etanercept's inhibition of ROS production, DNA damage, and downregulation of NF-κB/p53 signaling. Collectively, these findings establish a mechanistic link between TNF-α, NF-κB activation, and TSCs senescence, with therapeutic implications for Etanercept in tendon degeneration (Fig. 7).
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
HG and HC conceived the study, designed and performed experiments and wrote and edited the manuscript. QL designed and performed experiments. ZG and GF conceived the study, performed experiments and edited the manuscript. All authors have read and approved the final manuscript. ZG and GF confirm the authenticity of all the raw data.
Ethics approval and consent to participate
The present study was approved by Animal Experiment Center of Nantong University (approval no. S20230522-005).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the Chinese National Natural Science Foundation (grant nos. 82071838 and 82201983), the Science and Technology Project of Nantong City (grant nos. JC2021187 and MS22022104), Jiangsu Provincial Medical Key Discipline Cultivation Unit (grant no. JSDW202205), Jiangsu Provincial Research Hospital (grant no. YJXYY202204), The Second Level of the Sixth Jianghai Elite Training Project in Nantong City (grant no. 2022, III-602) and the 14th Five-Year Science and Education and Strong Health Engineering Medical Talents Program of Nantong City (grant no. 2021-2025).
References
|
Iagnocco A, Riente L, Delle Sedie A, Filippucci E, Salaffi F, Meenagh G, Scirè CA, Grassi W, Montecucco C, Bombardieri S and Valesini G: Ultrasound imaging for the rheumatologist. XXII. Achilles tendon involvement in spondyloarthritis. A multi-centre study using high frequency volumetric probe. Clin Exp Rheumatol. 27:547–551. 2009. | |
|
Alves EM, Macieira JC, Borba E, Chiuchetta FA and Santiago MB: Spontaneous tendon rupture in systemic lupus erythematosus: Association with Jaccoud's arthropathy. Lupus. 19:247–254. 2010. View Article : Google Scholar | |
|
Järvinen TA, Kannus P, Paavola M, Järvinen TL, Józsa L and Järvinen M: Achilles tendon injuries. Curr Opin Rheumatol. 13:150–155. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Lin YJ, Anzaghe M and Schülke S: Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells. 9:8802020. View Article : Google Scholar | |
|
Mobasheri A and Shakibaei M: Is tendinitis an inflammatory disease initiated and driven by pro-inflammatory cytokines such as interleukin 1β? Histol Histopathol. 28:955–964. 2013. | |
|
Sharma P and Maffulli N: Tendon injury and tendinopathy: Healing and repair. J Bone Joint Surg Am. 87:187–202. 2005. | |
|
Schulze-Tanzil G, Al-Sadi O, Wiegand E, Ertel W, Busch C, Kohl B and Pufe T: The role of pro-inflammatory and immunoregulatory cytokines in tendon healing and rupture: New insights. Scand J Med Sci Sports. 21:337–351. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Jeong C, Kim SE, Shim KS, Kim HJ, Song MH, Park K and Song HR: Exploring the in vivo anti-inflammatory actions of simvastatin-loaded porous microspheres on inflamed tenocytes in a collagenase-induced animal model of achilles tendinitis. Int J Mol Sci. 19:8202018. View Article : Google Scholar : PubMed/NCBI | |
|
Alfredson H, Lorentzon M, Bäckman S, Bäckman A and Lerner UH: cDNA-arrays and real-time quantitative PCR techniques in the investigation of chronic Achilles tendinosis. J Orthop Res. 21:970–975. 2003. View Article : Google Scholar | |
|
Choi S, Song MH, Shim KS, Kim HJ, Lim YM, Song HR, Park K and Kim SE: Therapeutic efficacy of intratendinous delivery of dexamethasone using porous microspheres for amelioration of inflammation and tendon degeneration on achilles tendinitis in rats. Biomed Res Int. 2020:50520282020. View Article : Google Scholar | |
|
Cho CH and Kim DH, Baek EH and Kim DH: Serum levels of TNF-α are increased in patients with rotator cuff tear and sleep disturbance. Diagnostics (Basel). 11:22152021. View Article : Google Scholar | |
|
Stengaard K, Hejbøl EK, Jensen PT, Degn M, Ta TML, Stensballe A, Andersen DC, Schroder HD, Lambertsen KL and Frich LH: Early-stage inflammation changes in supraspinatus muscle after rotator cuff tear. J Shoulder Elbow Surg. 31:1344–1356. 2022. View Article : Google Scholar | |
|
Frich LH, Fernandes LR, Schrøder HD, Hejbøl EK, Nielsen PV, Jørgensen PH, Stensballe A and Lambertsen KL: The inflammatory response of the supraspinatus muscle in rotator cuff tear conditions. J Shoulder Elbow Surg. 30:e261–e275. 2021. View Article : Google Scholar | |
|
Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, Jackson CG, Lange M and Burge DJ: A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 340:253–259. 1999. View Article : Google Scholar | |
|
Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, et al: Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 337:141–147. 1997. View Article : Google Scholar : PubMed/NCBI | |
|
Pennica D, Kohr WJ, Fendly BM, Shire SJ, Raab HE, Borchardt PE, Lewis M and Goeddel DV: Characterization of a recombinant extracellular domain of the type 1 tumor necrosis factor receptor: Evidence for tumor necrosis factor-alpha induced receptor aggregation. Biochemistry. 31:1134–1141. 1992. View Article : Google Scholar : PubMed/NCBI | |
|
Mease PJ, Goffe BS, Metz J, VanderStoep A, Finck B and Burge DJ: Etanercept in the treatment of psoriatic arthritis and psoriasis: A randomised trial. Lancet. 356:385–390. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Chen K, Li P, Zhao H, Yan X and Ma Y: Effects of tumor necrosis factor inhibitor on stress-shielded tendons. Orthopedics. 40:49–55. 2017. View Article : Google Scholar | |
|
Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW and Zoeller M: CD44 and EpCAM: Cancer-initiating cell markers. Curr Mol Med. 8:784–804. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Wang JHC and Komatsu I: Tendon stem cells: Mechanobiology and development of tendinopathy. Adv Exp Med Biol. 920:53–62. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang M, Liu H, Cui Q, Han P, Yang S, Shi M, Zhang T, Zhang Z and Li Z: Tendon stem cell-derived exosomes regulate inflammation and promote the high-quality healing of injured tendon. Stem Cell Res Ther. 11:4022020. View Article : Google Scholar : PubMed/NCBI | |
|
Yang Z, Cao H, Gao S, Yang M, Lyu J and Tang K: Effect of tendon stem cells in chitosan/β-glycerophosphate/collagen hydrogel on achilles tendon healing in a rat model. Med Sci Monit. 23:4633–4643. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, Lee SR and Yang SH: The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int J Mol Sci. 22:27192021. View Article : Google Scholar | |
|
Zhao Y, Zhu XY, Song T, Zhang L, Eirin A, Conley S, Tang H, Saadiq I, Jordan K, Lerman A and Lerman LO: Mesenchymal stem cells protect renal tubular cells via TSG-6 regulating macrophage function and phenotype switching. Am J Physiol Renal Physiol. 320:F454–F463. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Leone GM, Mangano K, Petralia MC, Nicoletti F and Fagone P: Past, present and (foreseeable) future of biological anti-TNF alpha therapy. J Clin Med. 12:16302023. View Article : Google Scholar : PubMed/NCBI | |
|
Mohamad Kamal NS, Safuan S, Shamsuddin S and Foroozandeh P: Aging of the cells: Insight into cellular senescence and detection methods. Eur J Cell Biol. 99:1511082020. View Article : Google Scholar : PubMed/NCBI | |
|
Perucca Orfei C, Lovati AB, Viganò M, Stanco D, Bottagisio M, Di Giancamillo A, Setti S and de Girolamo L: Dose-related and time-dependent development of collagenase-induced tendinopathy in rats. PLoS One. 11:e01615902016. View Article : Google Scholar : PubMed/NCBI | |
|
AVMA Guidelines for the Euthanasia of Animals: 2020 edition. Available from: https://www.avma.org/resources-tools/avmapolicies/avma-guidelines-euthanasia-animals.pdf. | |
|
Debacq-Chainiaux F, Erusalimsky JD, Campisi J and Toussaint O: Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 4:1798–1806. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Hernandez-Segura A, Nehme J and Demaria M: Hallmarks of cellular senescence. Trends Cell Biol. 28:436–453. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Qi Z, Yang W, Xue B, Chen T, Lu X, Zhang R, Li Z, Zhao X, Zhang Y, Han F, et al: ROS-mediated lysosomal membrane permeabilization and autophagy inhibition regulate bleomycin-induced cellular senescence. Autophagy. 20:2000–2016. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Gaida JE, Alfredson H, Forsgren S and Cook JL: A pilot study on biomarkers for tendinopathy: Lower levels of serum TNF-α and other cytokines in females but not males with achilles tendinopathy. BMC Sports Sci Med Rehabil. 8:52016. View Article : Google Scholar | |
|
Fredberg U and Ostgaard R: Effect of ultrasound-guided, peritendinous injections of adalimumab and anakinra in chronic Achilles tendinopathy: A pilot study. Scand J Med Sci Sports. 19:338–344. 2009. View Article : Google Scholar | |
|
Kemoun G and Defebvre L: Gait disorders in Parkinson disease. Clinical description, analysis of posture, initiation of stabilized gait. Presse Med. 30:452–459. 2001.In French. PubMed/NCBI | |
|
Gulotta LV, Kovacevic D, Cordasco F and Rodeo SA: Evaluation of tumor necrosis factor α blockade on early tendon-to-bone healing in a rat rotator cuff repair model. Arthroscopy. 27:1351–1357. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Kwan KHL, Yeung KWK, Liu X, Wong KKY, Shum HC, Lam YW, Cheng SH, Cheung KMC and To MKT: Silver nanoparticles alter proteoglycan expression in the promotion of tendon repair. Nanomedicine. 10:1375–1383. 2014. View Article : Google Scholar | |
|
Yang J, He J and Yang L: Advanced glycation end products impair the repair of injured tendon: A study in rats. BMC Musculoskelet Disord. 25:7002024. View Article : Google Scholar : PubMed/NCBI | |
|
Bi Y, Ehirchiou D, Kilts TM, Inkson CA, Embree MC, Sonoyama W, Li L, Leet AI, Seo BM, Zhang L, et al: Identification of tendon stem/progenitor cells and the role of the extracellular matrix in their niche. Nat Med. 13:1219–1227. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Shang D, Liu H and Tu Z: Pro-inflammatory cytokines mediating senescence of vascular endothelial cells in atherosclerosis. Fundam Clin Pharmacol. 37:928–936. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Kojima H, Kunimoto H, Inoue T and Nakajima K: The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle. 11:730–739. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, Herbert BS, Rajashekhar G, Ingram DA, Yoder MC, Clauss M and Rehman J: Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. FASEB J. 23:1358–1365. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Leppkes M, Roulis M, Neurath MF, Kollias G and Becker C: Pleiotropic functions of TNF-α in the regulation of the intestinal epithelial response to inflammation. Int Immunol. 26:509–515. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Jacobson EC, Jain L, Vickers MH, Olins AL, Olins DE, Perry JK and O'Sullivan JM: TNF-α differentially regulates cell cycle genes in promyelocytic and granulocytic HL-60/S4 cells. G3 (Bethesda). 9:2775–2786. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Huang Y, Zuo F, Wu J and Wu S: TNF-α regulated bidirectional interaction between bone marrow mesenchymal stem cells and articular chondrocytes. Cartilage. Nov 20–2024.Epub ahead of print. View Article : Google Scholar | |
|
Zhu L, Dissanayaka WL, Green DW and Zhang C: Stimulation of EphB2/ephrin-B1 signalling by tumour necrosis factor alpha in human dental pulp stem cells. Cell Prolif. 48:231–238. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Li W, Liu Q, Shi J, Xu X and Xu J: The role of TNF-α in the fate regulation and functional reprogramming of mesenchymal stem cells in an inflammatory microenvironment. Front Immunol. 14:10748632023. View Article : Google Scholar | |
|
Fan P, Yu XY, Chen CH, Gao JW, Xu YZ, Xie XH and Wang YT: Parkin-mediated mitophagy protects against TNF-α-induced stress in bone marrow mesenchymal stem cells. Exp Gerontol. 164:1118292022. View Article : Google Scholar | |
|
Jung YH, Chae CW, Chang HS, Choi GE, Lee HJ and Han HJ: Silencing SIRT5 induces the senescence of UCB-MSCs exposed to TNF-α by reduction of fatty acid β-oxidation and anti-oxidation. Free Radic Biol Med. 192:1–12. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou Z, Akinbiyi T, Xu L, Ramcharan M, Leong DJ, Ros SJ, Colvin AC, Schaffler MB, Majeska RJ, Flatow EL and Sun HB: Tendon-derived stem/progenitor cell aging: Defective self-renewal and altered fate. Aging Cell. 9:911–915. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Tan Q, Lui PPY and Rui YF: Effect of in vitro passaging on the stem cell-related properties of tendon-derived stem cells-implications in tissue engineering. Stem Cells Dev. 21:790–800. 2012. View Article : Google Scholar | |
|
Nie D, Zhang J, Zhou Y, Sun J, Wang W and Wang JHC: Rapamycin treatment of tendon stem/progenitor cells reduces cellular senescence by upregulating autophagy. Stem Cells Int. 2021:66382492021. View Article : Google Scholar : PubMed/NCBI | |
|
Boyce BF, Yao Z and Xing L: Functions of nuclear factor kappaB in bone. Ann N Y Acad Sci. 1192:367–375. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Alharbi KS, Fuloria NK, Fuloria S, Rahman SB, Al-Malki WH, Javed Shaikh MA, Thangavelu L, Singh SK, Rama Raju Allam VS, Jha NK, et al: Nuclear factor-kappa B and its role in inflammatory lung disease. Chem Biol Interact. 345:1095682021. View Article : Google Scholar : PubMed/NCBI | |
|
Bashiri Dezfouli A, Salar-Amoli J, Pourfathollah AA, Yazdi M, Nikougoftar-Zarif M, Khosravi M and Hassan J: Doxorubicin-induced senescence through NF-κB affected by the age of mouse mesenchymal stem cells. J Cell Physiol. 235:2336–2349. 2020. View Article : Google Scholar | |
|
Wang C, Zhou Z, Song W, Cai Z, Ding Z, Chen D, Xia F and He Y: Inhibition of IKKβ/NF-κB signaling facilitates tendinopathy healing by rejuvenating inflamm-aging induced tendon-derived stem/progenitor cell senescence. Mol Ther Nucleic Acids. 27:562–576. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Abraham AC, Shah SA, Golman M, Song L, Li X, Kurtaliaj I, Akbar M, Millar NL, Abu-Amer Y, Galatz LM and Thomopoulos S: Targeting the NF-kappaB signaling pathway in chronic tendon disease. Sci Transl Med. 11:eaav43192019. View Article : Google Scholar | |
|
Best KT, Nichols AEC, Knapp E, Hammert WC, Ketonis C, Jonason JH, Awad HA and Loiselle AE: NF-κB activation persists into the remodeling phase of tendon healing and promotes myofibroblast survival. Sci Signal. 13:eabb72092020. View Article : Google Scholar | |
|
Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, Fischkoff SA and Chartash EK: Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: A randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 50:1400–1411. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Genovese MC, Bathon JM, Martin RW, Fleischmann RM, Tesser JR, Schiff MH, Keystone EC, Wasko MC, Moreland LW, Weaver AL, et al: Etanercept versus methotrexate in patients with early rheumatoid arthritis: Two-year radiographic and clinical outcomes. Arthritis Rheum. 46:1443–1450. 2002. View Article : Google Scholar : PubMed/NCBI |










