



SMC2 as a potential prognostic biomarker in lung adenocarcinoma and its correlation with immune microenvironment
- Authors:
- Published online on: August 12, 2025 https://doi.org/10.3892/mco.2025.2888
- Article Number: 93
-
Copyright: © Zheng et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Lung cancer is among the most common malignant neoplasms with the worse prognosis globally. It is classified into two categories: Non-small cell lung cancer (NSCLC; ~85%) and small cell lung cancer (~15%) (1). Lung adenocarcinoma (LUAD) is the predominant histological subtype of NSCLC, representing ~60% of cases (2), with a 5-year survival rate of 22.1% (3). Novel treatments for NSCLC have been developed, encompassing surgery, chemotherapy, radiation, targeted therapy and immunotherapy (4). Nonetheless, the recurrence and mortality rates of LUAD remain elevated, resulting in a poor prognosis for patients. Consequently, it is essential to identify more efficacious biomarkers to enable novel therapeutic interventions.
Structural maintenance of chromosomes (SMC) proteins, a class of DNA-binding ATPases, are essential for preserving chromosomal cohesion during cell division (5). SMC denotes structural maintenance of chromosome complexes, comprising chromatin loop-extruding cohesin (extrusive cohesin), sister chromatid cohesive cohesin and mitotic chromosome-associated condensation (6). SMC2 is one of the six members of the SMC family, forming a dimer with SMC4, which binds to three non-SMC proteins to form a pentameric complex called Condensin (7). Condensin has DNA supercoiling activity, essential for chromatin compaction prior to cell division, and it has been demonstrated to be essential for the disintegration of sister chromatids in subsequent phases (8,9). Thus, SMC2 proteins serve an important role in the organization of chromatin packaging prior to cell division and the DNA damage response, which is needed for the maintenance of chromosome stability (10). Moreover, previous research has reported that they may have pro-carcinogenic functions, with SMC2 knockdown inhibiting tumor growth in colorectal cancer and SMC2 involvement in cell mitosis (7). In addition, experimental studies have reported that SMC2 knockdown increased apoptosis in neuroblastoma cells (10). Badea et al (11) reported that the expression level of SMC2 mRNA was markedly higher in human pancreatic cancer tissues than in adjacent normal tissues. Meanwhile, several other genes of the SMC family have been reported to be closely associated with tumorigenesis, such as SMC1A (12), SMC3(13) and SMC4(14). To a certain extent, this supports the connection between SMC2 and cancer aggressiveness.
The present study utilized RNA sequencing data from The Cancer Genome Atlas (TCGA) database and other bioinformatics analysis methods, including the Human Protein Atlas (HPA) and University of ALabama at Birmingham CANcer data analysis Portal (UALCAN), to assess SMC2 mRNA expression in LUAD tissues, and these results were further evaluated using cell lines and tissues. Secondly, Gene Set Enrichment Analysis (GSEA) was utilized to assess its possible mechanisms of action in LUAD. The cBioPortal database was also employed to evaluate SMC2 mutations in LUAD. The Tumor Immune Estimation Resource (TIMER) and single-sample (ss)GSEA algorithms were used to assess the association between SMC2 expression and immune cell infiltration in LUAD. Finally, the association between SMC2 and immunological checkpoints, chemokines and receptors was evaluated utilizing the Tumor-Immune System Interaction Database (TISIDB). Collectively, the findings of the present study offer novel insights into the prognosis, mutation and immune response of SMC2 in LUAD.
Materials and methods
Ethical statement
The present study was performed in accordance with the Declaration of Helsinki, and the protocol was approved (approval no. 2023-187) by the Ethics Committee of Hunan Provincial People's Hospital (Changsha, China). Moreover, 18 pairs of fresh LUAD and adjacent non-tumor tissues were collected for analysis. The Ethics Committee authorized the use of these samples, and all patients participating in the trial provided informed consent.
Access to RNA sequencing data and clinical information
The sequencing data and corresponding clinicopathologic information for 539 LUAD samples and 59 adjacent normal samples were downloaded from the TCGA database (https://portal.gdc.cancer.gov/) (15). To compensate for the relative lack of normal samples when performing differential gene expression analysis, SMC2 expression data was obtained from 347 normal lung tissue samples sourced from the Genotype-Tissue Expression (GTEx) database (https://www.gtexportal.org/) (16).
Reverse transcription-quantitative polymerase chain reaction analysis (RT-qPCR)
Total RNA was extracted from cell lines (BEAS-2B, A549, H1975 and PC9) as well as from 18 pairs of LUAD tissues using TRIzol (Jiangsu Kangwei Century Biotechnology Co., Ltd.). Subsequently, RT-qPCR was performed using a cDNA synthesis kit (Qiagen, Inc.) and a SYBR real-time PCR kit (Qiagen, Inc.) in accordance with the manufacturers' protocols. Quantitative analysis was performed using the 2-ΔΔCq method (17). The sequences of the SMC2 primers were as follows: forward, 5'-TCTCAGGTTCGGGCTTCTAAT-3' and reverse, 5'-CCTGTACTCTGGTGTTGTTGG-3'. The internal reference gene was β-actin, in which the forward sequence was 5'-TCTCCCAAGTCCACACAGG-3' and the reverse, 5'-GGCACGAAGGCTCATCA-3'. Thermocycling conditions were as follows: The initial denaturation time was 15 min at 95˚C; followed by a denaturation time of 15 sec at 94˚C, an annealing time of 30 sec at 55˚C, and an extension time of 30 sec at 72˚C. The number of cycles for denaturation, annealing, and extension was 40 cycles.
Immunohistochemistry (IHC)
Initially, fresh tissue samples were fixed in 4% paraformaldehyde at 4˚C for 24 h, dried using ethanol, embedded in paraffin and subsequently sectioned. Sections (5 µm) underwent deparaffinization using xylene, rehydration in 100, 95, 85, and 75% alcohol, respectively, for 5 min each, immersion in sodium citrate and microwave treatment for 15 min for antigen extraction. Sections were treated overnight at 4˚C with the anti-SMC2 rabbit/IgG primary antibodies (cat. no. 84835-1-RR; Proteintech Group, Inc.) at a dilution of 1:100. Sections were incubated at 37˚C for 30 min with secondary antibody goat anti-mouse/rabbit IgG (cat. no. PV-6000, supplied by Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.). Sections were stained using diaminobenzidine and subsequently counterstained with hematoxylin at 25˚C and for 1 min. Ultimately, images were captured using a Zeiss light microscope (Zeiss GmbH) and assessed using with ImageJ software (1.53K; National Institutes of Health).
UALCAN and HPA databases
UALCAN (http://ualcan.path.uab.edu/) is an interactive web resource that is comprehensive and user-friendly, allowing users to recognize biomarkers or validate potential genes of interest in silico (18). In the present study, it was used to assess the protein expression levels of SMC2 at different levels. In addition, IHC data for SMC2 expression was obtained from the HPA (19).
Nomogram construction and evaluation
Indexes possessing independent prognostic significance identified from multivariate Cox analyses were incorporated into Nomogram models forecasting 1-, 3- and 5-year disease-specific survival (DSS) in patients with LUAD. Predictive efficacy was evaluated using calibration curves generated using the ‘rms’ R software package.
Protein-protein interaction (PPI)
Initially, gene correlation analysis was performed on SMC2, followed by the identification of differentially expressed genes (DEGs) with |log2FoldChange| >1.2 in LUAD to generate volcano plots. Subsequently, the top 800 genes and DEGs most closely associated with SMC2 in the TCGA database were analyzed using Venn diagrams, and the identified target molecules were utilized for subsequent PPI and Gene Ontology (GO) analyses. The PPI network was constructed in the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://cn.string-db.org/) (20) and then imported into Cytoscape (version 3.10.0; https://cytoscape.org/release_notes_3_10_0.html) for adjustments to find molecules with a greater associated with SMC2 in LUAD.
GO and GSEA
DEGs associated with SMC2 were utilized for GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses to assess the biological processes in which SMC2 may participate in LUAD. GO analysis was employed to assess the biological processes, molecular functions and cellular components of the mRNAs. GSEA is an approach for route enrichment analysis that effectively manages type I and type II errors, making it widely utilized in the study of multi-omics data (21). The analysis utilized the ‘clusterprofiler’ in the R package (https://github.com/YuLab-SMU/clusterProfiler). A total of 1,000 operations were performed in each analysis, with normalized enrichment scores of >1, false discovery rates of <0.25 and adjusted P<0.05 was considered to indicate a statistically significant difference.
Genetic alterations in the SMC2 gene in LUAD
The distribution of different types of SMC2 mutations in cancer were analyzed using the Catalogue of Somatic Mutations in Cancer (COSMIC) website (https://cancer.sanger.ac.uk/cosmic/) (22). Moreover, the distribution of SMC2 mutations in cancer were analyzed using three datasets of LUAD from the cBioPortal (http://www.cbioportal.org/) platform (23, 24) (TCGA, Firehose Legacy; TCGA, Nature 2014; TCGA, PanCancer Atlas) to evaluate SMC2 gene alterations in LUAD.
ssGSEA and TIMER 2.0
ssGSEA is an enhancement of the GSEA methodology that computes enrichment scores for each sample and gene set combination. Thus, ssGSEA transforms gene expression profiles of individual samples into gene set enrichment profiles. Consequently, ssGESA was employed to assess the association between SMC2 and several tumor-infiltrating immune cells. TIMER 2.0(25) (http://timer.comp-genomics.org/) is a comprehensive resource that systematically evaluates the association between genes and the prevalence of immune-infiltrating cells across several cancer types, allowing users to assess the complete spectrum of immunological, clinical and genomic characteristics of tumors. The present study employed these data to evaluate the relationship between SMC2 and the prevalence of immune-infiltrating cells within the tumor microenvironment (TME).
TISIDB databases
TISIDB (http://cis.hku.hk/TISIDB/) (26) serves as a platform for assessing interactions between tumors and the immune system. This platform was used to systematically evaluate the association between SMC2 and immune checkpoint inhibitors, stimulators, chemokines and receptors in LUAD.
Statistical analysis
All statistical analyses and statistical graphics were performed with R software (version 3.6.3; https://cran.r-project.org/src/base/R-3/). Analysis of variance (ANOVA) is used to compare differences in SMC2 expression among multiple groups, supplemented by the Bonferroni post-hoc test. The Z-test or Wilcoxon rank-sum test is used for comparisons between two groups. Using the chi-squared test or Fisher's exact test to analyze the relationship between SMC2 expression and clinical pathological features. The ‘pROC software’ package was utilized to generate receiver operating characteristic (ROC) curves, whilst Kaplan-Meier followed by the log-rank test and Cox analyses evaluated the impact of SMC2 on the survival outcomes of patients with LUAD. Spearman correlation analysis was performed to elucidate the relationship between SMC2 expression and tumor-infiltrating lymphocytes (TILs), as well as immune correlations involving checkpoint inhibitors, chemokines and related genes. P<0.05 was considered to indicate a statistically significant difference.
Results
Pan-cancer analysis of SMC2 mRNA and upregulation of expression in LUAD
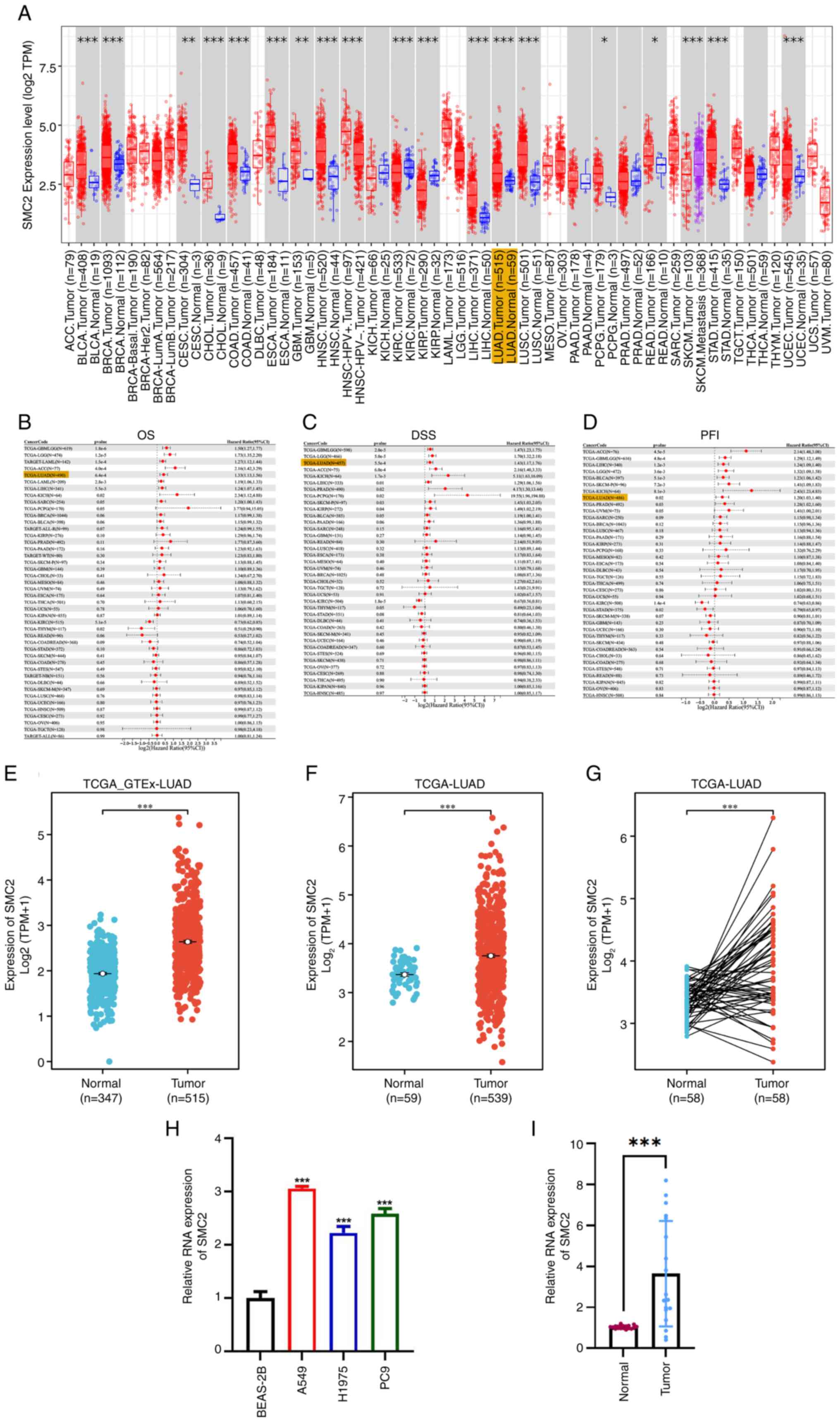
Considering the function of SMC2 in several human malignancies, the present study performed a pan-cancer assessment of SMC2 mRNA expression utilizing the TCGA database. The findings indicated that SMC2 had elevated expression in several malignancies, including LUAD, uterine corpus endometrial carcinoma and colon adenocarcinoma (Fig. 1A). Subsequently, a thorough analysis of the impact of SMC2 on overall survival (OS), DSS and progression-free interval (PFI) in patients with cancer was performed, revealing that the overexpression of SMC2 may negatively affect the prognosis of patients with LUAD (Fig. 1B-D). Consequently, LUAD was selected as the cancer of focus in the investigation. The results demonstrated that the expression levels of SMC2 are elevated in LUAD tissues, irrespective of the incorporation of data from the GTEx database (Fig. 1E and F). This tendency was similarly observed in matched samples (Fig. 1G). Ultimately, elevated expression of SMC2 was confirmed in LUAD cell lines (A549, H1975 and PC9) and human bronchial epithelial cells BEAS-2B, in addition to 18 pairs of LUAD tissues and adjacent normal tissues from Hunan Provincial People's Hospital (Fig. 1H and I). Collectively, the findings indicate that SMC2 is significantly expressed in LUAD.
Expression of SMC at the protein level in LUAD
Using the Clinical Proteomic Tumor Analysis Consortium database of the UALCAN platform, it was revealed that the protein expression level of SMC2 in LUAD was increased relative to normal tissues (Fig. 2A). Subsequently, the SMC2 protein expression levels in LUAD were derived from IHC staining data in the HPA database (antibody no. HPA071309). Consistent with the upregulation of mRNA levels, SMC2 protein expression was significantly upregulated in LUAD tissues (patient ID nos. 1907, 2003 and 4923) relative to normal tissues (patient ID nos. 1470, 1678 and 2643) (Fig. 2B). Furthermore, protein expression levels of SMC2 were progressively higher in pathological grades two and three compared with the normal group, although there was no significant difference for grade one (Fig. 2C). Alterations in the mTOR and HIPPO pathways combined with the Wnt pathway were also associated with elevated SMC2 protein expression levels (Fig. 2D-F). Furthermore, the expression of SMC2 was assessed using IHC in 70 LUAD tissues from Hunan Provincial People's Hospital (Fig. S1).

Association between SMC2 and clinicopathologic features
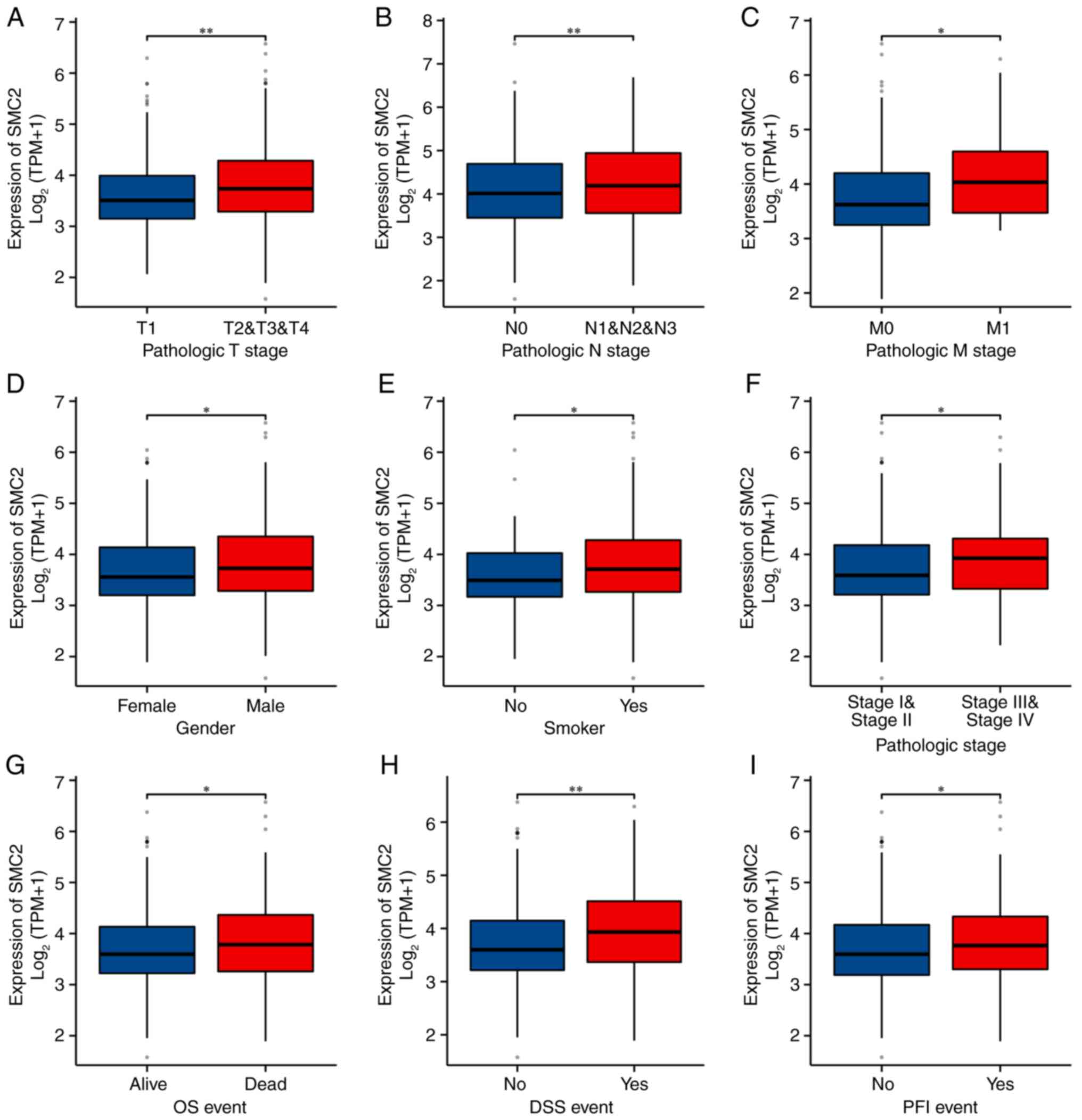
The results demonstrated that SMC2 was significantly overexpressed in patients with LUAD with tumor (T)2-T4 stage, node (N)1-N3 stage, metastasis (M)1 stage, higher pathologic stage and OS fatal event, along with in patients with DSS/PFI events (Fig. 3A-I). Therefore, it was hypothesized that SMC2 may adversely influence the survival of patients.
Diagnostic and prognostic value of SMC2 in LUAD
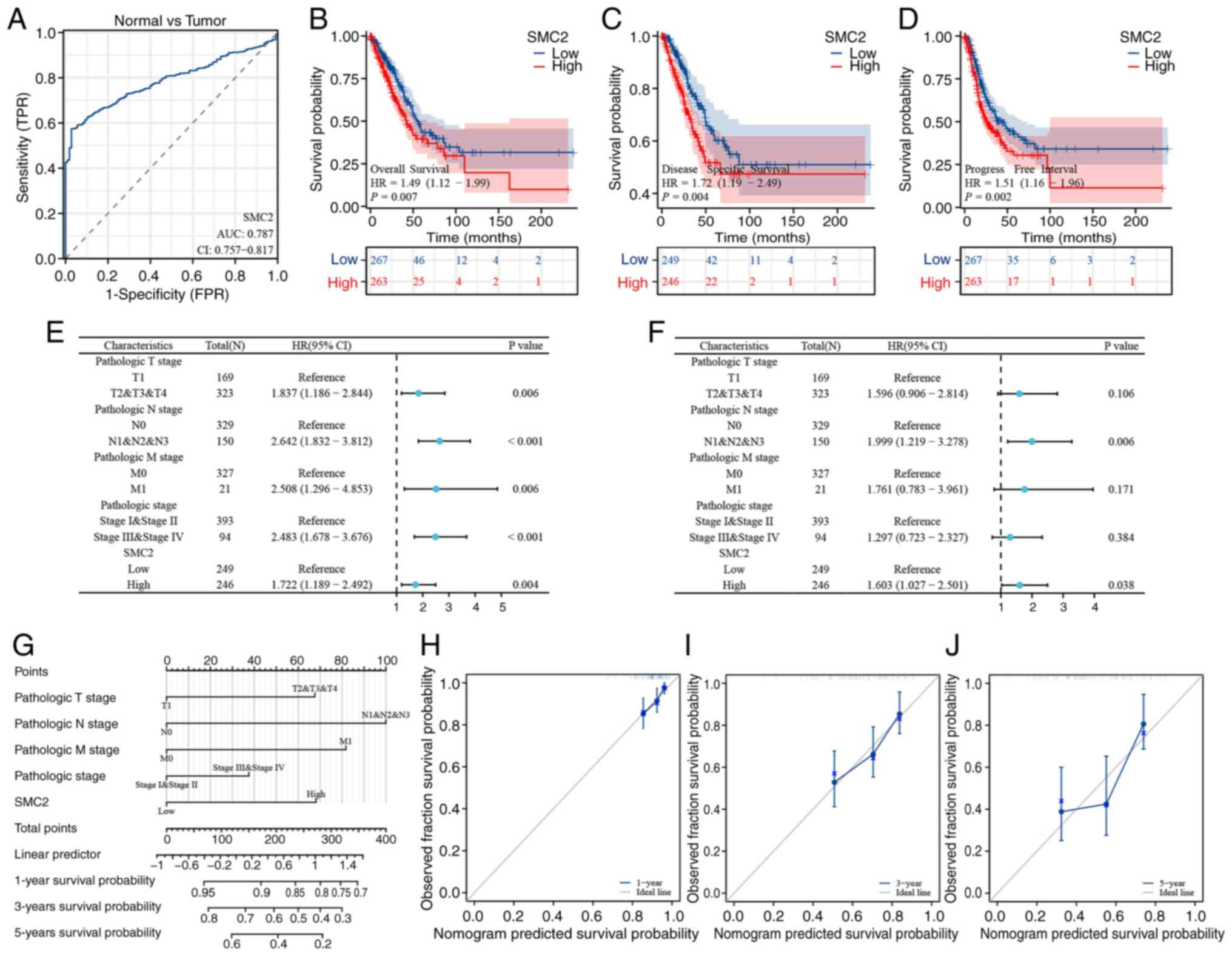
The diagnostic-effect of SMC2 in LUAD was first assessed using ROC curves. The results demonstrated that the AUC value of SMC2 in distinguishing LUAD tissues from normal tissues was 0.787 (Fig. 4A). Moreover, Kaplan-Meier curves revealed the effect of SMC2 expression on the tumor prognosis of patients with LUAD, with the SMC2 high-expression group significantly associated with a worse OS, DSS and PFI in patients with LUAD, compared with the SMC2 low-expression group. The hazard ratios for OS, DSS and PFI were 1.49, 1.72 and 1.51, respectively (Fig. 4B-D; P<0.05). Furthermore, univariate (Fig. 4E) and multivariate (Fig. 4F) Cox regression analyses were performed, which further demonstrated that SMC2 may be an independent prognostic factor for LUAD. In addition, according to the results of Cox analysis, a nomogram was constructed to predict the DSS of patients with LUAD at 1, 3 and 5 years (Fig. 4G). Notably, the C-index for evaluating its predictive effect was 0.701, and the calibration curve demonstrated a comparatively favorable agreement between the predicted and actual values (Fig. 4H-J).
In addition, the clinical data of 70 patients with LUAD were obtained from Hunan Provincial People's Hospital (Table SI). Based on the expression of SMC2 detected by IHC, the patients were divided into high- and low-expression groups, accounting for 54.3 and 45.7%, respectively. The results revealed that high expression of SMC2 was associated with pathological grade and T stage. Moreover, the OS of patients in the high expression group of SMC2 was poor (Fig. S2).
Filtering mRNA potentially relevant to SMC2
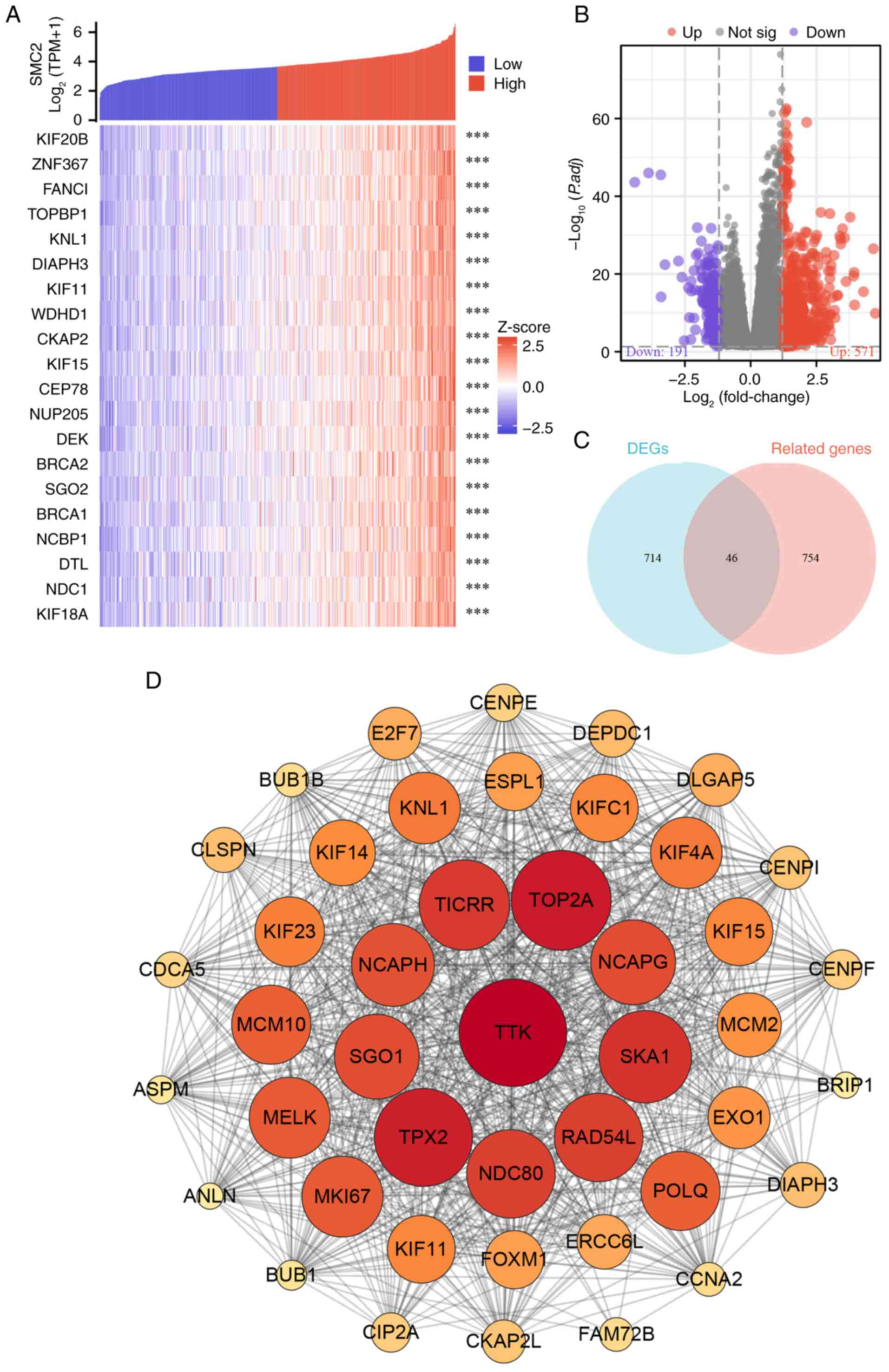
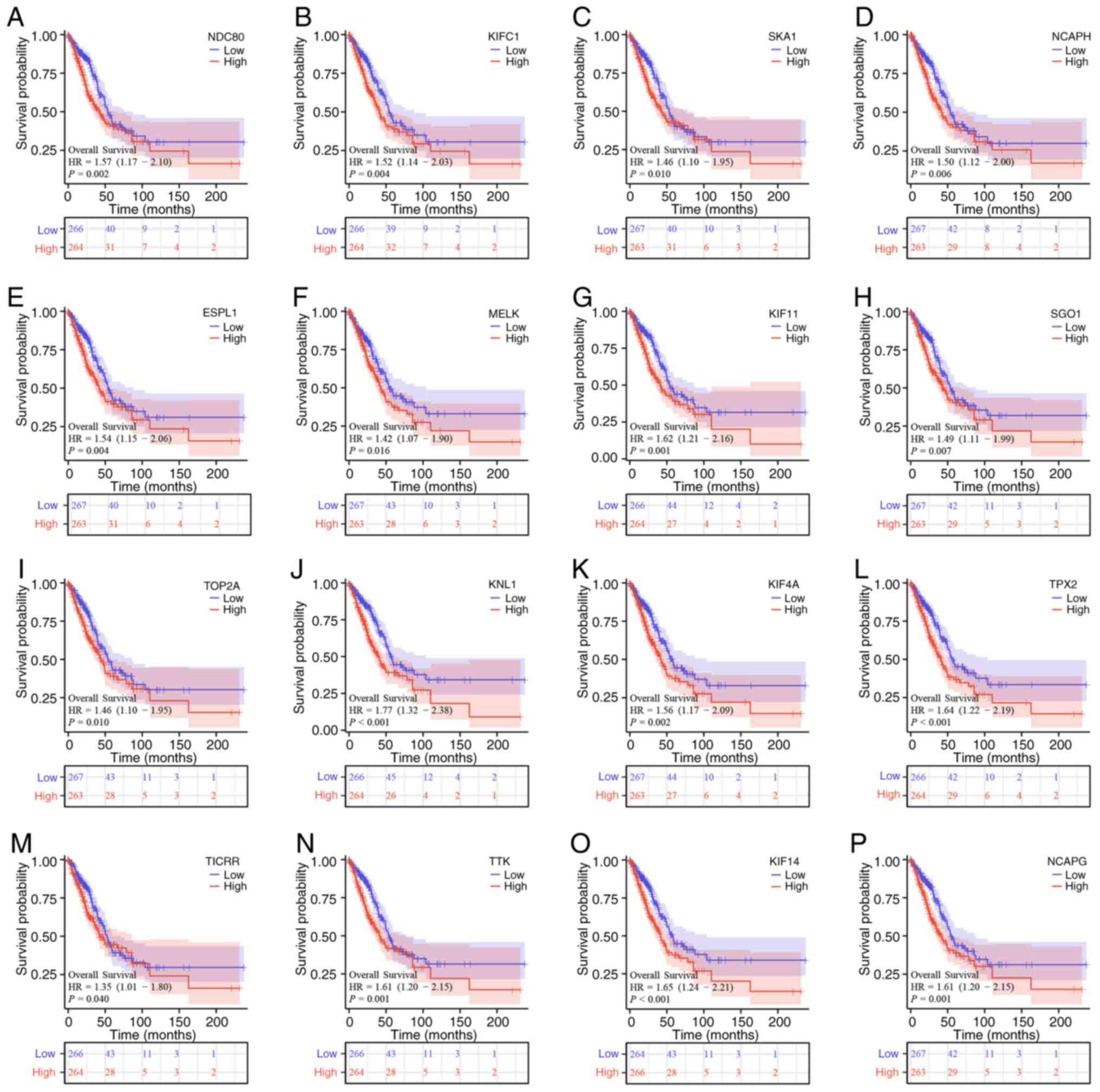
The top 800 mRNAs that were closely associated with SMC2 in LUAD were first selected and the generated heatmap revealed the top 20 molecules (Fig. 5A). Subsequently, differentially expressed mRNAs in LUAD were screened by volcano plots according to the filtering standard, in which 750 mRNAs were either up- or downregulated (Fig. 5B). Venn diagrams further identified overlapping genes in the two groups (Fig. 5C). These 46 target molecules were then constructed into a PPI network using the STRING database, and the hub genes were selected based on the centrality of the nodes (Fig. 5D). Finally, Kaplan-Meier survival analysis of mRNAs (NDC80, KIFC1, SKA1, NCAPH, ESPL1, MELK, KIF11, SGO1, TOP2A, KNL1, KIF4A, TPX2, TICRR, TTK, KIF14 and NCAPG) was performed to further assess their effects on OS in patients with LUAD. Notably, upregulation of all these molecules in LUAD exacerbated the poor prognosis of OS in patients (Fig. 6A-P), and the detrimental effect of these mRNAs on the survival prognosis of patients with LUAD is consistent with the tendency of SMC2.
Exploring the potential mechanisms of SMC2 in LUAD
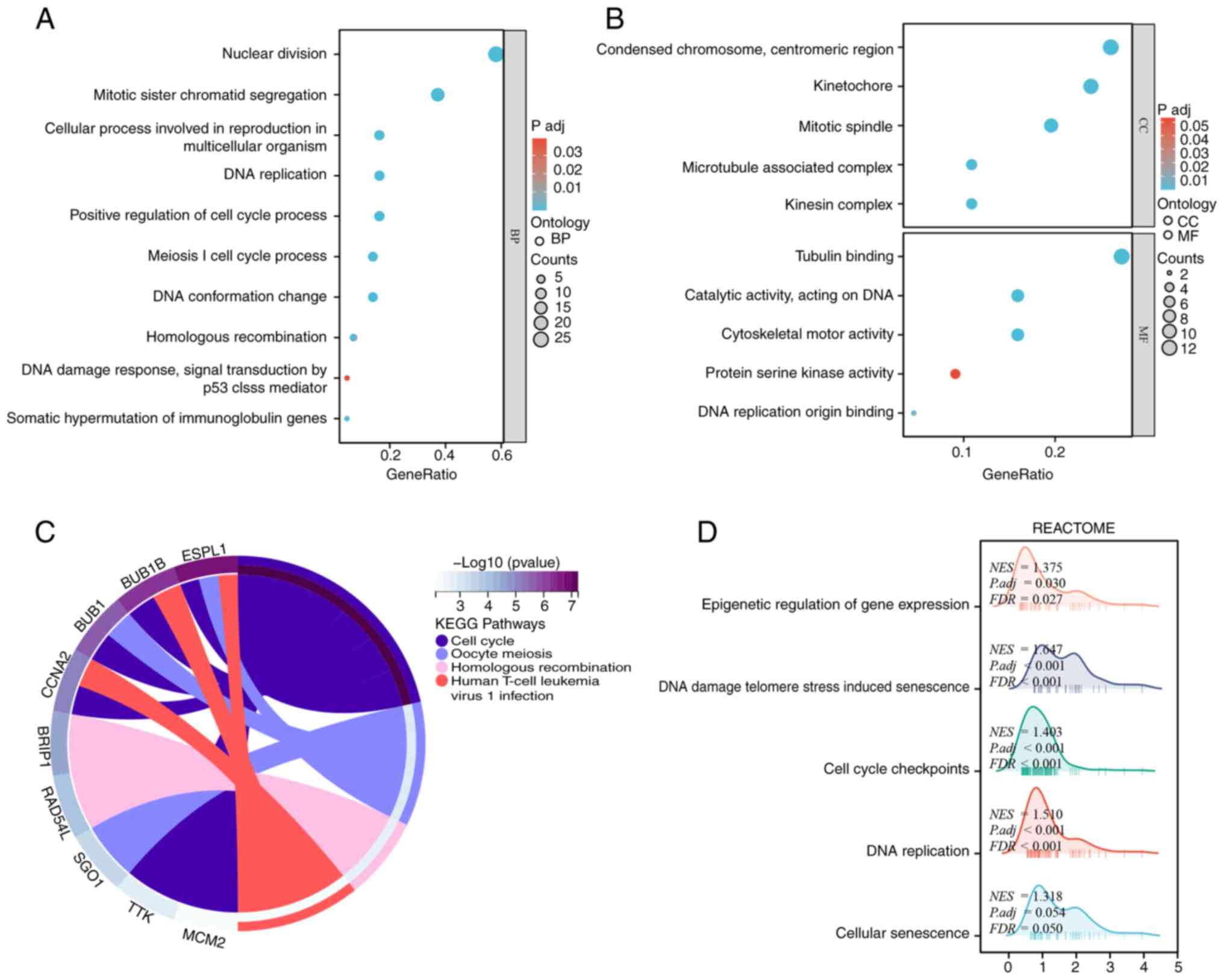
Based on the 46 targets mRNA obtained from the pre-screening, GO analysis was performed which revealed that SMC2 may be involved in the following biological processes: nuclear division, mitotic sister chromatid segregation, cellular process involved in reproduction in multicellular organism and DNA replication. Meanwhile, it was demonstrated that SMC2 is involved in cellular components such as condensed chromosome, centromeric region, kinetochore and mitotic spindle, and its potential molecular function is mainly to influence tubulin binding (Fig. 7A and B). In addition, the potential pathway of SMC2 in LUAD was evaluated using GSEA. The results revealed that the significantly enriched KEGG pathways included cell cycle, oocyte meiosis, homologous recombination and human T-cell leukemia virus 1 infection. The REACTOME pathways included DNA damage telomere stress induced senescence, DNA replication, cell cycle checkpoints, epigenetic regulation of gene expression as well as cellular senescence (Fig. 7C and D). These findings may provide a reference for further studies in the future.
SMC2 genetic alteration in LUAD
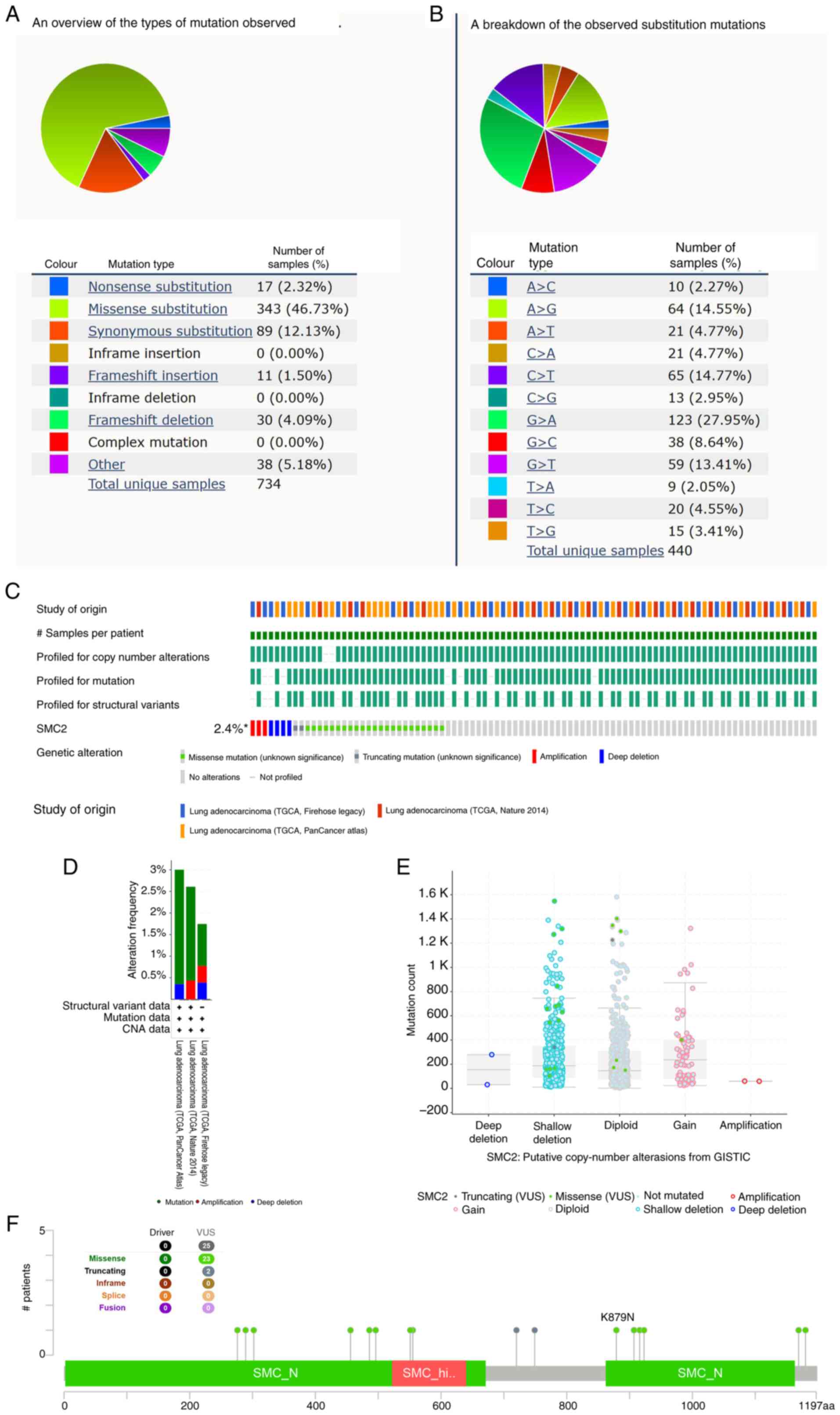
The COSMIC website revealed the distribution of different types of SMC2 mutations in cancer (Fig. 8A and B). Missense substitutions were the most common type of mutation, following by synonymous substitutions, and G>A, C>T and A>G were the most common substitution mutations. Subsequently, the SMC2 gene mutations in LUAD were analyzed using three datasets in the cBioPortal database. The results indicated that SMC2 was genetically altered in 2.4% of patients with LUAD (Fig. 8C). Moreover, the specific types and frequency of gene mutations were visualized (Fig. 8D). SMC2 mRNA expression was elevated in the shallow deletion group compared with in the diploid group and notably, the expression of gain groups was also increased (Fig. 8E). In the LUAD samples, 23 SMC2 missense mutation sites were identified, one of which was K879N, suggesting that this is one of the protein activation hotspots. A total of two SMC2 truncating mutation sites were also identified (Fig. 8F).
Association between SMC2 expression and immune characteristics in LUAD
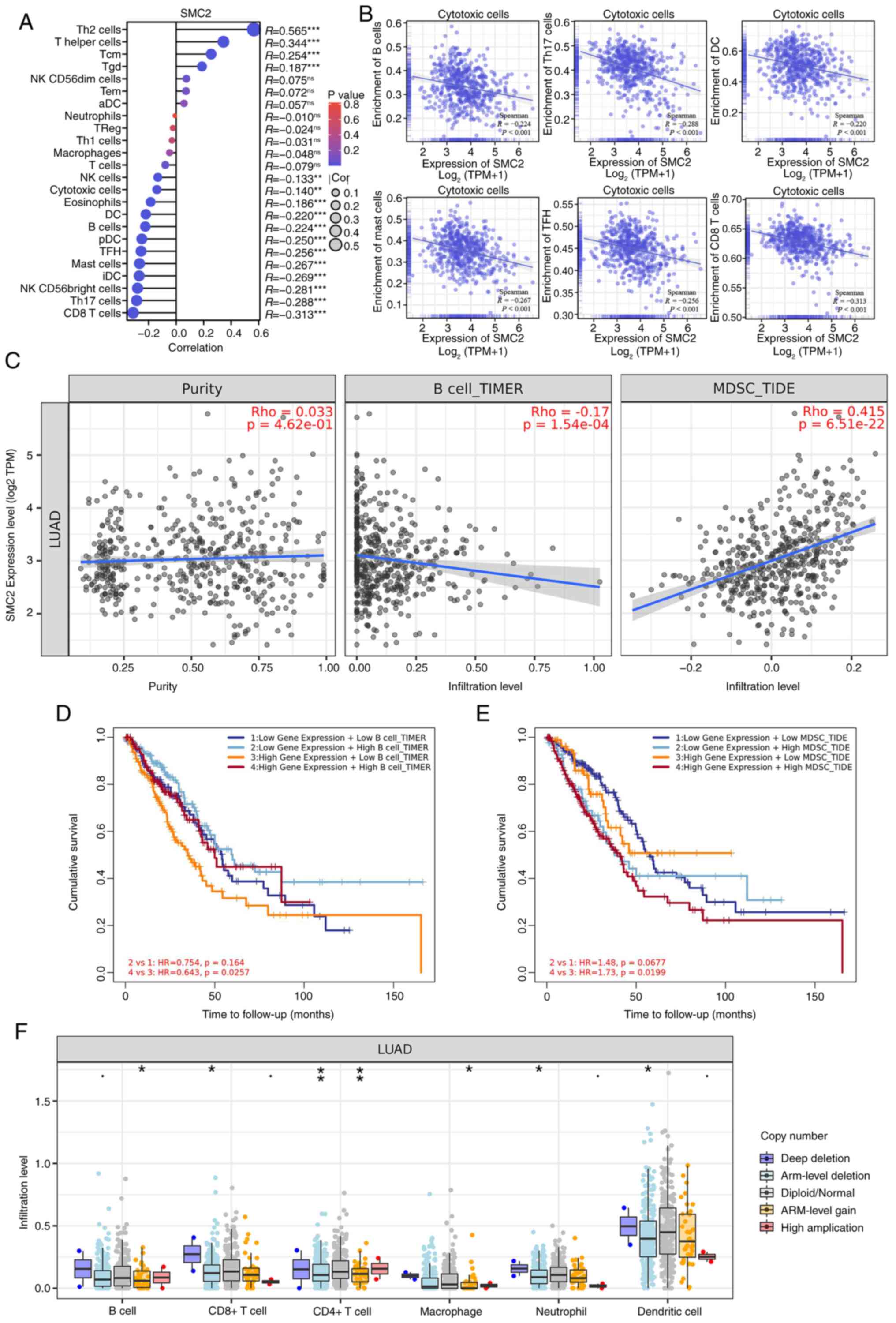
To further assess the role of SMC2 in LUAD, the correlation between SMC2 and TILs was analyzed. Based on the ssGSEA algorithm, it was demonstrated that SMC2 is negatively correlated with CD8+ T cells, Th 17 cells, mast cells, dendritic cells, T follicular helper cells (TFH) and B cells (Fig. 9A and B). By contrast, using the TIMER and the tumor immune dysfunction and exclusion (TIDE) algorithms in the TIMER database, it was revealed that SMC2 is negatively and positively correlated with activated B cells and myeloid-derived suppressor cells (MDSCs), respectively (Fig. 9C). Notably, when activated B cells were enriched, the OS of patients with LUAD was significantly improved; however, when MDSCs were enriched, the OS of patients with LUAD was dramatically worse (Fig. 9D and E). From the aforementioned results, it was hypothesized that the cancer-promoting effects of SMC2 may be related to a certain extent to the low enrichment of B cells as well as the high enrichment of MDSCs. Furthermore, the findings indicated that the arm-level deletion of SMC2 copy number was associated with reduced abundance of CD8+ T cells, CD4+ T cells, neutrophils and dendritic cells when compared with the diploid/normal state (Fig. 9F). These results indicate that copy number changes of SMC2 in LUAD may be an element that regulates the immune microenvironment.
Immune checkpoints serve a vital role in the immune microenvironment of LUAD, and these directly regulate the antitumor immune response (27). Therefore, the present study analyzed the association between SMC2 and immuno-inhibitors (Fig. 10A). Notably, SMC2 was significantly positively correlated with CD274 (r=0.298; P<6.26x10-12) and PDCD1LG2 (r=0.226; P<2.29x10-7) (Fig. 10B). The association between SMC2 and an immuno-stimulator was also analyzed (Fig. 10D). Furthermore, SMC2 was significantly negatively correlated with TNFSF13 (r=-0.471; P<2.2x10-16), TMEM173 (r=-0.404; P<2.2x10-16), TNFRSF14 (r=-0.399; P<2.2x10-16) and HHLA2 (r=-0.282; P<7.53x10-11) (Fig. 10C).
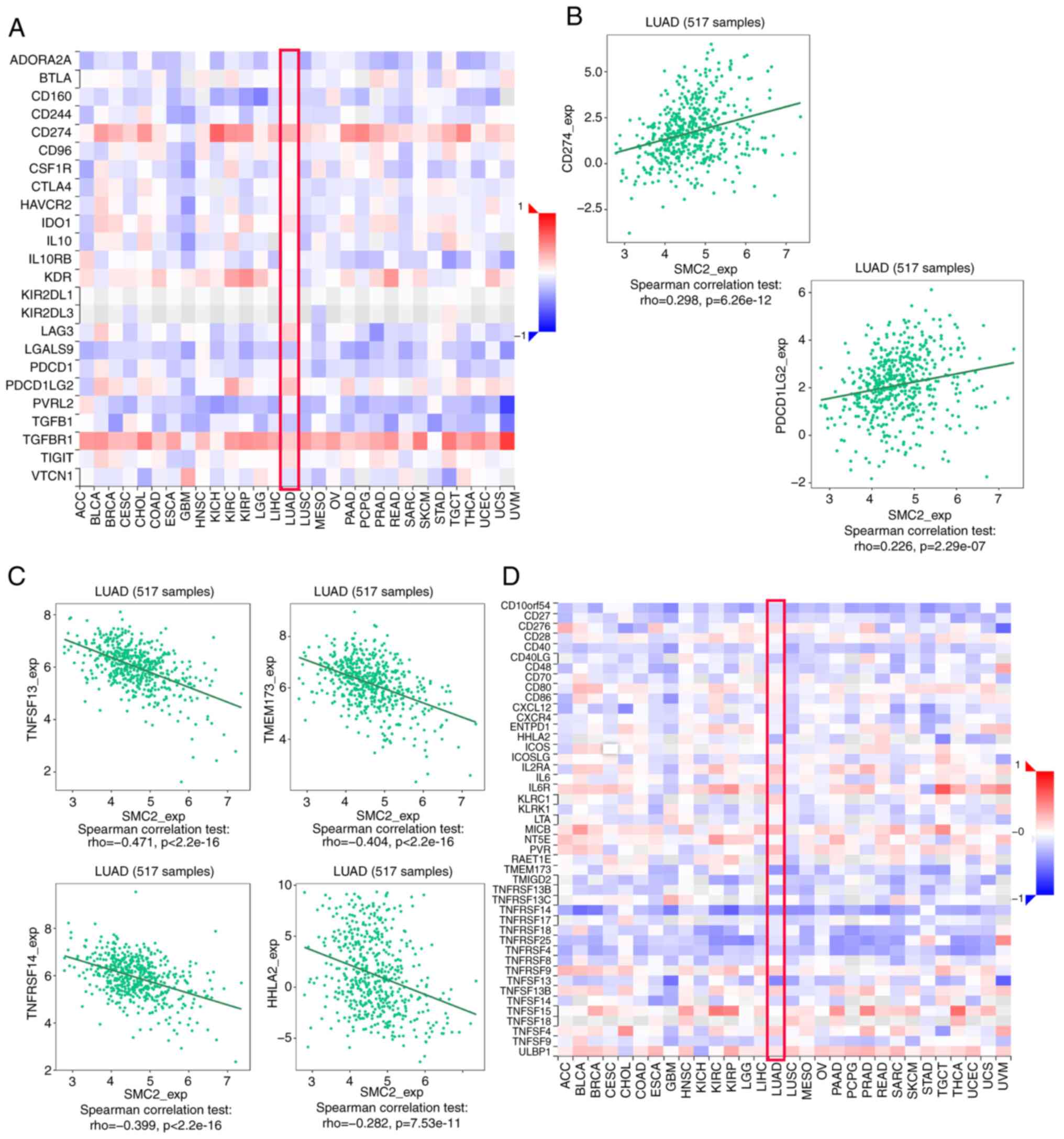
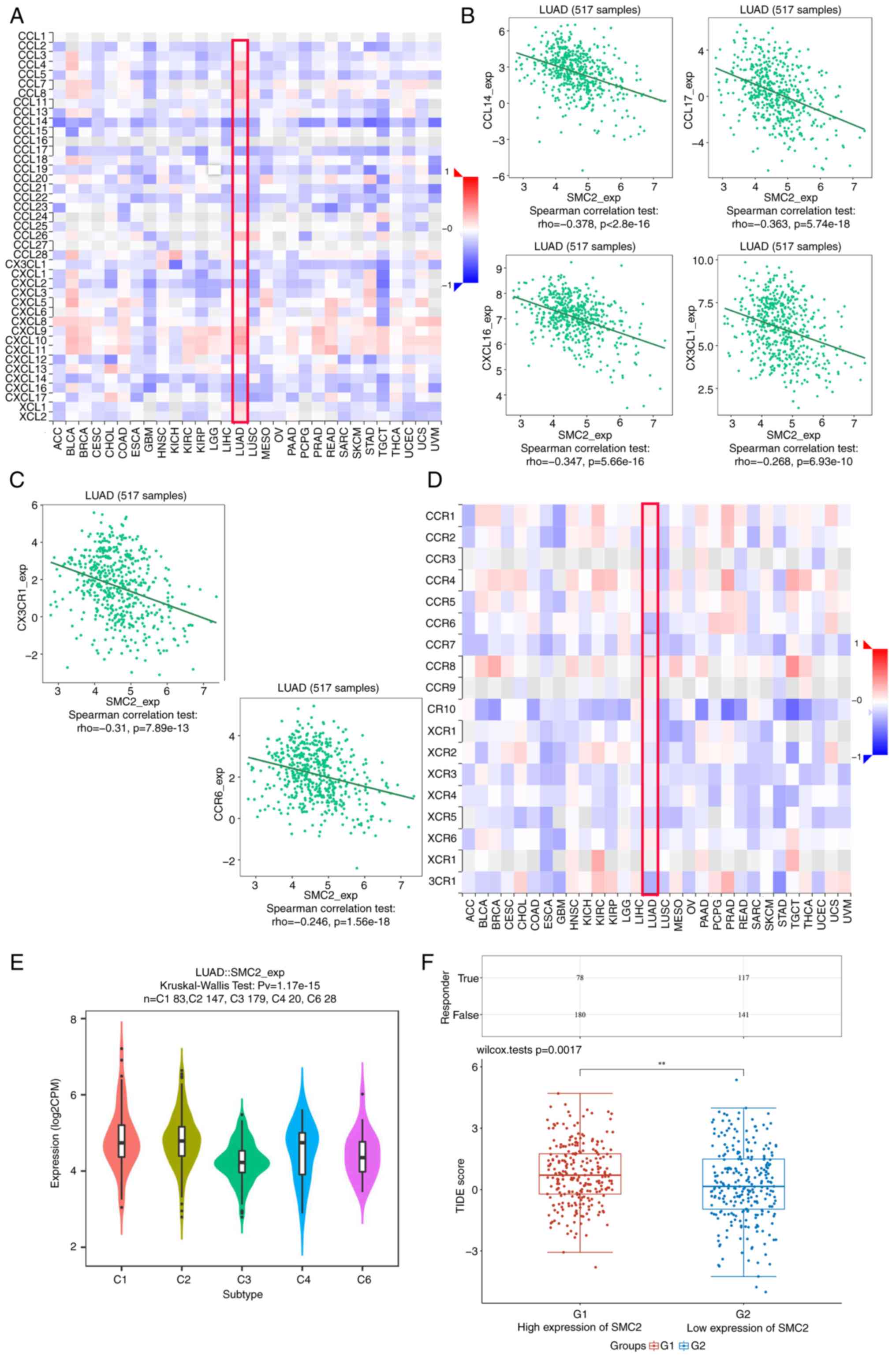
Chemokines and chemokine receptors are essential for tumor infiltration by immune cells (28). Therefore, the present study analyzed the correlation between SMC2 expression levels and immune cell chemokines and receptors in LUAD using the TISIDB database. Heatmap results revealed that several chemokines and receptors were significantly correlated with SMC2 expression in LUAD (Fig. 11A and D). Subsequently, the correlation between SMC2 expression and chemokines/receptors was analyzed. The results demonstrated that SMC2 expression was significantly negatively correlated with CCL14 (r=-0.378; P<2.2x10-16), CCL17 (r=-0.363; P<5.74x10-18), CXCL16 (r=-0.347; P<5.66x10-16), CX3CL1 (r=-0.268; P<6.93x10-10), CX3CR1 (r=-0.31; P<7.89x10-13) and CCR6 (r=-0.246; P<1.56x10-8) (Fig. 11B and C). These results suggest that the SMC2 gene may serve an influential role in tumor immune environment. In addition, the relationship between SMC2 and LUAD immune subtypes was analyzed. The findings revealed that SMC2 was highly expressed in type C2 (IFN-γ-dominant) and type C4 (lymphocyte-depleted) immune subtypes, whilst it was least expressed in type C3 (inflammatory) (Fig. 11E). This suggests that the expression of SMC2 may be associated with the immune microenvironment of LUAD. Finally, the TIDE algorithm was used, which is extensively used to predict cancer immunotherapy response, with higher TIDE scores associated with worse immunotherapy outcomes (29). The results revealed that high SMC2 expressors had significantly higher TIDE scores compared with low expressors, which resulted in a lower response rate to immunotherapy (Fig. 11F).
Discussion
The SMC family comprises prokaryotic and eukaryotic chromosomal proteins that are essential for the organization of chromosomal structure. SMC2 is the second member of the SMC family in budding yeast and encompasses all the presumed structural domains typical of SMC family members: A nucleotide-binding region, two coiled-coil regions divided by a ‘hinge’, and the carboxy-terminus of Smclp (DA box) itself. The SMC2 protein has been reported to be a subunit of the human condensing complex (8). Previous research indicates that the SMC family is associated with several human malignancies, including pancreatic, hepatocellular and colorectal cancers (30-33). SMC2 belongs to the condensin complex, which has also been reported to be associated with apoptosis in neuroblastoma cells (10). In the present study, SMC2 was demonstrated to be a potential prognostic biomarker for LUAD using biological databases and the cohort from Hunan Provincial People's Hospital. The present study first observed high expression of SMC2 in LUAD tissue from 8 patients with LUAD, followed by the collection of 10 additional samples for validation. These findings provide preliminary evidence that SMC2 may serve as a potential prognostic biomarker for LUAD. Nevertheless, the authors acknowledged that the overall sample size of the present study remains limited. Therefore, while the findings of the present study are significant, they should be interpreted with caution and validated through larger-scale, multi-center studies to ensure the reliability and generalizability of the results. SMC2 expression was elevated in LUAD samples, revealing a beneficial diagnostic impact on LUAD, and it was associated with worse OS, DSS and PFI in patients with LUAD. These findings align with previous studies (30-33), suggesting that SMC2 may serve as a promising biological marker for LUAD. To further evaluate the clinical value of SMC2 as a prognostic biomarker, the present study compared its prognostic ability with known LUAD gene biomarkers (such as KRAS, TP53 and EGFR). First, in univariate Cox regression analysis, the present study found that the expression of SMC2 and KRAS was significantly associated with poorer survival outcomes (P<0.05), while TP53 and EGFR mutations were not significantly associated with survival outcomes (P>0.05) (Table SII). Second, in multivariate Cox regression analysis, the present study found that SMC2 was not significantly associated with KRAS, TP53, or EGFR mutations in terms of survival outcomes. This suggests that the prognostic value of SMC2 may be independent of these known LUAD biomarkers.
The molecular interactions within the LUAD TME constitute a complex network. The present study identified several important proteins potentially associated with SMC2 using correlation analysis and PPI network design, such as TTK, TOP2A, TICRR, NCAPH, SKA1, TPX2, NDC80 and SGO1 (34-37). Notably, these molecules have been reported to be associated with the malignant phenotype and a worse prognosis of LUAD in previous studies. These data not only provide a possible regulation network, but also indirectly validate the malicious function of SMC2 in LUAD. Genetic alterations are closely associated with cancer and several genetic alterations have been reported to be associated with the occurrence and development of LUAD (38,39). The present study demonstrated that missense substitutions were the most common type of mutation followed by synonymous substitutions in 734 samples. G>A (27.95%), C>T (14.77%) and A>G (14.55%) were the most common substitution mutations. Furthermore, the frequency of SMC2 gene mutation was only 2.4% in LUAD, which was mainly missense mutations. Thus, exploring the mechanism by which mutation affects the prognosis and therapeutic response of patients with LUAD could provide further benefits to the patients.
Similar to other members of the SMC family, SMC2 is considered to participate in cell cycle regulation (40). The present study demonstrated that the results of GO, KEGG and GSEA analysis corroborate the relevance of SMC2 in LUAD. Tumor formation is associated with the TME. The TME comprises immune cells, extracellular matrix, mesenchymal stromal cells and inflammatory mediators, all of which influence tumor growth, metastasis and clinical survival outcomes (41). Prior research has indicated that immune infiltration may affect patient prognosis, and the grading of TILs serves as an independent predictor of sentinel lymph node status in patients with tumors (42). B cells are identified as the TILs most strongly associated with risk scores, particularly in individuals with metastatic LUAD (43). Research has reported that CD8+ T lymphocytes can identify and eliminate cancer cells (44), and CD8+ T cells have been a focal point in clinical cancer therapy for >20 years for the detection and elimination of malignant cells (45). Guo et al (46) identified circulating TFH cells that may serve an essential role in the development of NSCLC pathogenesis. Notably, the present study demonstrated that SMC2 expression was negatively correlated with the level of infiltration of these cells. This is consistent with previous findings, suggesting to a certain extent that tumorigenesis may be associated with the absence or low enrichment of lymphocytes, which further contributes to the malignant progression of the disease. In addition, the low enrichment of B cells in LUAD was associated with the deterioration of OS (Fig. 9D), which also further corroborates the results of ssGSEA with TIMER. MDSCs are immature myeloid cells that promote tumor growth and metastasis by inducing immunosuppression, which ultimately affects patient outcomes (47). In the present study, SMC2 expression was positively correlated with the level of MDSC infiltration, and highly enriched MDSC led to a worse prognosis (Fig. 9E). Therefore, SMC2 may inhibit the function of immune cells by increasing the level of MDSC infiltration, thus becoming a potential immunotherapeutic target.
Systemic treatment options for patients with advanced LUAD have been markedly expanded to include not only chemotherapy and targeted therapy, but also immune checkpoint inhibitors (27). The present study demonstrated that SMC2 was positively correlated with immune checkpoints such as CD274 and PDCD1LG2 in LUAD. This suggests that SMC2 may synergize with immune checkpoints, thus further exacerbating the immunosuppressive state in the TME.
Moreover, previous studies have reported that chemokines can directly or indirectly modulate the TME and biological phenotype, influencing angiogenesis, tumorigenesis and malignant metastasis, among other processes (48). In the present study, the relationship between SMC2 expression levels and the expression of chemokines and receptors in LUAD was assessed utilizing the TISIDB database. The findings indicated that the expression level of SMC2 exhibited a negative correlation with the expression of CCL14, CCL17, CXCL16, CX3CL1, CX3CR1 and CCR6, suggesting that elevated SMC2 expression may impede the migration of immune cells to the TME. CCL17, also known as thymus and activation-regulated chemokine, is a C-C chemokine often associated with type 2 immune responses (49), and CCR6 controls integrin-mediated adhesion in B cells (50,51). The CXCR5+ subpopulation of CD8+ T cells may contribute to antitumor activity (52), and the receptor for CX3CL1 and CX3CR1, in turn, controls leukocyte survival and natural killer cell activation (53). The robust connection of SMC2 with these molecules may elucidate its role in regulating immune infiltration in LUAD, suggesting that its interaction with chemokines in tumors could also be a contributing element to the malignant character of tumors. The TIDE algorithm was also utilized to evaluate the responsiveness of patients across several SMC2 expression groups to immune checkpoint treatment. The TIDE ratings of the SMC2 high-expression group were considerably elevated compared with those of the SMC2 low-expression group, indicating that the immunotherapy response rate may be diminished in patients within the SMC2 high-expression group. This suggests that the simultaneous inhibition of SMC2 and immunological checkpoints may represent a viable method for the treatment of LUAD in the near future.
Currently, insight into the mechanism of SMC2 in the tumor immune microenvironment is lacking. Future studies should elucidate in more detail how SMC2 affects immune cell infiltration, immune checkpoint expression and tumor cell immunogenicity. For example, key immune-related genes regulated by SMC2 need to be identified and the interaction of SMC2 with other immune signaling pathways should be explored. Alternatively, inhibition of SMC2 may enhance the sensitivity of tumors to immune checkpoint inhibitors, thereby improving therapeutic efficacy. Only when there is a full understanding of the mechanism of action of SMC2 can SMC2-targeted therapeutic strategies be developed in a more targeted manner. For example, instead of directly inhibiting SMC2 expression, inhibitors that target specific functions of SMC2 could be developed. In addition, the interactions of SMC2 with other proteins could be explored to identify new therapeutic targets. More precise SMC2 assays are also required to improve the assessment of SMC2 expression levels in tumor tissues. This will help to identify patients who are most likely to benefit from SMC2-targeted therapy. In conclusion, translating SMC2 into clinical applications is a long-term process that requires notable research investment. Future studies should focus on elucidating the mechanism of action of SMC2, developing more accurate SMC2 assays and exploring more selective SMC2-targeted therapeutic strategies.
Moreover, the present study aimed to explore the potential clinical application value of SMC2 in LUAD through bioinformatics analysis. Using publicly available datasets, the present study found that SMC2 is highly expressed in LUAD, and high expression is associated with poorer survival rates, suggesting that SMC2 may serve as a potential prognostic biomarker for LUAD. To validate these bioinformatics findings, the present study conducted qPCR validation in cell lines and LUAD tissue samples, and the validation results were consistent with the bioinformatics analysis results, providing support for the conclusions of the present study. However, the present study also has certain limitations. First, the present study primarily relied on bioinformatics analysis and qPCR validation, lacking direct functional experiments such as SMC2 knockdown or overexpression, as well as proliferation, migration and invasion experiments to directly assess its impact on LUAD cell behavior. Secondly, the precise mechanism of SMC2 in LUAD carcinogenesis and progression remains ambiguous; the present study has not yet fully explored the specific mechanism of action of SMC2 in LUAD. Although bioinformatic analysis suggests that SMC2 may be associated with certain signaling pathways, the present study did not experimentally verify whether these pathways are regulated by SMC2, nor did it elucidate the specific role of SMC2 in these pathways. Third, although the results of the present study demonstrated that SMC2 expression is intricately associated with immune infiltration and prognosis in LUAD, data demonstrating that SMC2 influences prognosis via its role in immune infiltration is lacking. The aforementioned limitations warrant additional investigation in the future. Given the aforementioned limitations, future research directions should include functional validation, mechanism studies, and in vivo experiments. The present study demonstrated that current bioinformatics analysis and preliminary validation provide a valuable starting point for understanding the potential role of SMC2 in LUAD. Future functional studies will be able to more comprehensively elucidate the specific mechanisms of SMC2 in LUAD progression, providing a basis for developing new treatment strategies and ultimately improving the prognosis of patients with LUAD.
In conclusion, to the best of the authors' knowledge, the present study presented the first systematic evidence for SMC2 in LUAD. The results revealed that SMC2 expression is upregulated in LUAD and is associated with a poor clinical prognosis of patients with LUAD. In addition, SMC2 expression was negatively correlated with inhibitory TILs, immuno-stimulators, chemokines and receptors. Moreover, its crosstalk with these factors may contribute to the malignant phenotype of LUAD and may provide a potential therapeutic target for patients with LUAD.
Supplementary Material
SMC2 expression in lung adenocarcinoma was assessed by IHC (magnification, x200). The cohort was divided subsets with low or high expression of SMC2 with immunohistochemistry score. SMC2, structural maintenance of chromosomes 2.
Survival curves of overall survival between patients with lung adenocarcinoma with either high or low expression of SMC2. SMC2, structural maintenance of chromosomes 2; HR, hazard ratio.
Relationship between SMC2 expression and clinicopathological variables.
Univariate and Multivariate Cox analysis of SMC2 and other prognostic biomarkers.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Ren-Shu Fund of Hunan Provincial People's Hospital (Grant No. RS201819) and the Natural Science Foundation of Hunan Province (Grant No. 2025 JJ80761).
Availability of data and materials
The data generated in the present study are included in the figures and/or tables of this article.
Authors' contributions
XT and FZ designed the study. YL, HC and YP were responsible for the statistical analysis. FZ wrote and plotted the manuscript, and performed the experiments. XT reviewed and revised the manuscript. FZ and XT confirm the authenticity of all the raw data. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
All experiments involving human tissues complied with the principles of the Declaration of Helsinki. The patient data in the present study, both from public databases and studies involving human subjects, was reviewed and approved by the Ethics Committee of Hunan Provincial People's Hospital (approval no. 2023-187; Changsha, China). All patients/participants provided written informed consent to participate in the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Siegel RL, Miller KD and Jemal A: Cancer statistics, 2020. CA Cancer J Clin. 70:7–30. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Denisenko TV, Budkevich IN and Zhivotovsky B: Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 9(117)2018.PubMed/NCBI View Article : Google Scholar | |
|
Kim HC, Jung CY, Cho DG, Jeon JH, Lee JE, Ahn JS, Kim SJ, Kim Y, Kim YC, Kim JE, et al: Clinical characteristics and prognostic factors of lung cancer in Korea: A pilot study of data from the Korean nationwide lung cancer registry. Tuberc Respir Dis (Seoul). 82:118–125. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Herbst RS, Morgensztern D and Boshoff C: The biology and management of non-small cell lung cancer. Nature. 553:446–454. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Hirano T: At the heart of the chromosome: SMC proteins in action. Nat Rev Mol Cell Biol. 7:311–322. 2006.PubMed/NCBI View Article : Google Scholar | |
|
Zhao H, Shu L, Qin S, Lyu F, Liu F, Lin E, Xia S, Wang B, Wang M, Shan F, et al: Extensive mutual influences of SMC complexes shape 3D genome folding. Nature. 640:543–553. 2025.PubMed/NCBI View Article : Google Scholar | |
|
Dávalos V, Súarez-López L, Castaño J, Messent A, Abasolo I, Fernandez Y, Guerra-Moreno A, Espín E, Armengol M, Musulen E, et al: Human SMC2 protein, a core subunit of human condensin complex, is a novel transcriptional target of the WNT signaling pathway and a new therapeutic target. J Biol Chem. 287:43472–43481. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Hudson DF, Marshall KM and Earnshaw WC: Condensin: Architect of mitotic chromosomes. Chromosome Res. 17:131–144. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Paliulis LV and Nicklas RB: Micromanipulation of chromosomes reveals that cohesion release during cell division is gradual and does not require tension. Curr Biol. 14:2124–2129. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Murakami-Tonami Y, Kishida S, Takeuchi I, Katou Y, Maris JM, Ichikawa H, Kondo Y, Sekido Y, Shirahige K, Murakami H and Kadomatsu K: Inactivation of SMC2 shows a synergistic lethal response in MYCN-amplified neuroblastoma cells. Cell Cycle. 13:1115–1131. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Badea L, Herlea V, Dima SO, Dumitrascu T and Popescu I: Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 55:2016–2027. 2008.PubMed/NCBI | |
|
Yadav S, Kowolik CM, Lin M, Zuro D, Hui SK, Riggs AD and Horne DA: SMC1A is associated with radioresistance in prostate cancer and acts by regulating epithelial-mesenchymal transition and cancer stem-like properties. Mol Carcinog. 58:113–125. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Kraft B, Lombard J, Kirsch M, Wuchter P, Bugert P, Hielscher T, Blank N and Krämer A: SMC3 protein levels impact on karyotype and outcome in acute myeloid leukemia. Leukemia. 33:795–799. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Jiang L, Zhou J, Zhong D, Zhou Y, Zhang W, Wu W, Zhao Z, Wang W, Xu W, He L, et al: Overexpression of SMC4 activates TGFβ/Smad signaling and promotes aggressive phenotype in glioma cells. Oncogenesis. 6(e301)2017.PubMed/NCBI View Article : Google Scholar | |
|
Tomczak K, Czerwińska P and Wiznerowicz M: The cancer genome atlas (TCGA): An immeasurable source of knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI View Article : Google Scholar | |
|
GTEx Consortium. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 348:648–660. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.PubMed/NCBI View Article : Google Scholar | |
|
Chandrashekar DS, Karthikeyan SK, Korla PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne U, et al: UALCAN: An update to the integrated cancer data analysis platform. Neoplasia. 25:18–27. 2022.PubMed/NCBI View Article : Google Scholar | |
|
Asplund A, Edqvist PH, Schwenk JM and Pontén F: Antibodies for profiling the human proteome-The human protein atlas as a resource for cancer research. Proteomics. 12:2067–2077. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al: The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49:D605–D612. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Canzler S and Hackermüller J: multiGSEA: A GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinformatics. 21(561)2020.PubMed/NCBI View Article : Google Scholar | |
|
Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR and Wooster R: The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 91:355–358. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al: The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2:401–404. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al: Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 6(pl1)2013.PubMed/NCBI View Article : Google Scholar | |
|
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B and Liu XS: TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48:W509–W514. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Ru B, Wong CN, Tong Y, Zhong JY, Zhong SSW, Wu WC, Chu KC, Wong CY, Lau CY, Chen I, et al: TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics. 35:4200–4202. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Chi A, He X, Hou L, Nguyen NP, Zhu G, Cameron RB and Lee JM: Classification of non-small cell lung cancer's tumor immune micro-environment and strategies to augment its response to immune checkpoint blockade. Cancers (Basel). 13(2924)2021.PubMed/NCBI View Article : Google Scholar | |
|
Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T, Kane LP and Ferris RL: PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 75:508–518. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al: Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 24:1550–1558. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Feng Y, Liu H, Duan B, Liu Z, Abbruzzese J, Walsh KM, Zhang X and Wei Q: Potential functional variants in SMC2 and TP53 in the AURORA pathway genes and risk of pancreatic cancer. Carcinogenesis. 40:521–528. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Yan W, Wang DD, Zhang HD, Huang J, Hou JC, Yang SJ, Zhang J, Lu L and Zhang Q: Expression profile and prognostic values of SMC family members in HCC. Medicine (Baltimore). 101(e31336)2022.PubMed/NCBI View Article : Google Scholar | |
|
Nie H, Wang Y, Yang X, Liao Z, He X, Zhou J and Ou C: Clinical significance and integrative analysis of the SMC family in hepatocellular carcinoma. Front Med (Lausanne). 8(727965)2021.PubMed/NCBI View Article : Google Scholar | |
|
Je EM, Yoo NJ and Lee SH: Mutational and expressional analysis of SMC2 gene in gastric and colorectal cancers with microsatellite instability. APMIS. 122:499–504. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Xu Y, Wang S, Xu B, Lin H, Zhan N, Ren J, Song W, Han R, Cheng L, Zhang M and Zhang X: AURKA, TOP2A and MELK are the key genes identified by WGCNA for the pathogenesis of lung adenocarcinoma. Oncol Lett. 25(238)2023.PubMed/NCBI View Article : Google Scholar | |
|
Li C, Meng J and Zhang T: NCAPH is a prognostic biomarker and associated with immune infiltrates in lung adenocarcinoma. Sci Rep. 12(9578)2022.PubMed/NCBI View Article : Google Scholar | |
|
Chen C, Guo Q, Song Y, Xu G and Liu L: SKA1/2/3 serves as a biomarker for poor prognosis in human lung adenocarcinoma. Transl Lung Cancer Res. 9:218–231. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Zhou F, Wang M, Aibaidula M, Zhang Z, Aihemaiti A, Aili R, Chen H, Dong S, Wei W and Maimaitiaili A: TPX2 promotes metastasis and serves as a marker of poor prognosis in non-small cell lung cancer. Med Sci Monit. 26(e925147)2020.PubMed/NCBI View Article : Google Scholar | |
|
Ricciuti B, Arbour KC, Lin JJ, Vajdi A, Vokes N, Hong L, Zhang J, Tolstorukov MY, Li YY, Spurr LF, et al: Diminished efficacy of programmed death-(Ligand)1 inhibition in STK11- and KEAP1-mutant lung adenocarcinoma is affected by KRAS mutation status. J Thorac Oncol. 17:399–410. 2022.PubMed/NCBI View Article : Google Scholar | |
|
Wohlhieter CA, Richards AL, Uddin F, Hulton CH, Quintanal-Villalonga À, Martin A, de Stanchina E, Bhanot U, Asher M, Shah NS, et al: Concurrent mutations in STK11 and KEAP1 promote ferroptosis protection and SCD1 dependence in lung cancer. Cell Rep. 33(108444)2020.PubMed/NCBI View Article : Google Scholar | |
|
Thadani R, Kamenz J, Heeger S, Muñoz S and Uhlmann F: Cell-Cycle regulation of dynamic chromosome association of the condensin complex. Cell Rep. 23:2308–2317. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Wang M, Chang M, Li C, Chen Q, Hou Z, Xing B and Lin J: Tumor-microenvironment-activated reactive oxygen species amplifier for enzymatic cascade cancer starvation/chemodynamic/immunotherapy. Adv Mater. 34(e2106010)2022.PubMed/NCBI View Article : Google Scholar | |
|
Slack FJ and Chinnaiyan AM: The role of non-coding RNAs in oncology. Cell. 179:1033–1055. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Shao MM, Zhai K, Huang ZY, Yi FS, Zheng SC, Liu YL, Qiao X, Chen QY, Wang Z and Shi HZ: Characterization of the alternative splicing landscape in lung adenocarcinoma reveals novel prognosis signature associated with B cells. PLoS One. 18(e0279018)2023.PubMed/NCBI View Article : Google Scholar | |
|
van der Leun AM, Thommen DS and Schumacher TN: CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat Rev Cancer. 20:218–232. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Wang Y, Li Y, Jiang X, Gu Y, Zheng H, Wang X, Zhang H, Wu J and Cheng Y: OPA1 supports mitochondrial dynamics and immune evasion to CD8(+) T cell in lung adenocarcinoma. PeerJ. 10(e14543)2022.PubMed/NCBI View Article : Google Scholar | |
|
Guo Z, Liang H, Xu Y, Liu L, Ren X, Zhang S, Wei S and Xu P: The role of circulating T Follicular helper cells and regulatory cells in non-small cell lung cancer patients. Scand J Immunol. 86:107–112. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Kalathil SG and Thanavala Y: Importance of myeloid derived suppressor cells in cancer from a biomarker perspective. Cell Immunol. 361(104280)2021.PubMed/NCBI View Article : Google Scholar | |
|
Nagarsheth N, Wicha MS and Zou W: Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 17:559–572. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Islam SA and Luster AD: T cell homing to epithelial barriers in allergic disease. Nat Med. 18:705–715. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Zlotnik A and Yoshie O: The chemokine superfamily revisited. Immunity. 36:705–716. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Matsukawa A, Hogaboam CM, Lukacs NW, Lincoln PM, Evanoff HL and Kunkel SL: Pivotal role of the CC chemokine, macrophage-derived chemokine, in the innate immune response. J Immunol. 164:5362–5368. 2000.PubMed/NCBI View Article : Google Scholar | |
|
Xing J, Zhang C, Yang X, Wang S, Wang Z, Li X and Yu E: CXCR5+CD8+ T cells infiltrate the colorectal tumors and nearby lymph nodes, and are associated with enhanced IgG response in B cells. Exp Cell Res. 356:57–63. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Ness TL, Ewing JL, Hogaboam CM and Kunkel SL: CCR4 is a key modulator of innate immune responses. J Immunol. 177:7531–7539. 2006.PubMed/NCBI View Article : Google Scholar |










