



A novel defined manganese metabolism‑related gene signature for predicting the prognosis of pancreatic ductal adenocarcinoma
- Authors:
- Published online on: July 9, 2025 https://doi.org/10.3892/ol.2025.15182
- Article Number: 436
-
Copyright: © Xiong et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal and aggressive cancer types, ranking as the fourth leading cause of cancer-related deaths globally. The bleak outlook and elevated mortality rates associated with pancreatic cancer are chiefly due to its aggressive characteristics and the absence of efficient early detection techniques. In 2021, the United States recorded ~60,430 new instances of pancreatic cancer, resulting in ~48,220 deaths, highlighting its considerable impact on public health (1). The prevalence of PDAC is increasing, and it is anticipated to emerge as the second leading cause of cancer-related mortality in the United States by 2030, provided that existing trends continue (2).
PDAC is frequently identified at advanced stages, resulting in a scarcity of curative treatment options. A total of ~80% of individuals receive a diagnosis of locally advanced or metastatic pancreatic cancer, which makes surgical resection unattainable for most, despite it being the sole potential curative intervention (3). Additionally, individuals diagnosed with metastatic PDAC exhibit a median overall survival (OS) of <1 one year, whilst the 5-year survival rate is ~10% (4). The inherent resistance of PDAC to chemotherapeutic agents and radiation modalities complicates therapeutic interventions, as most patients encounter recurrence or disease progression within a few months following initial treatment (5).
The absence of effective early detection methods notably contributes to the high mortality rate. Most patients do not exhibit symptoms until the disease advances, and current imaging modalities, such as CT and MRI, lack sufficient sensitivity for early diagnosis (6). Although biomarkers such as cancer antigen 19-9 are recognized as potential diagnostic and prognostic tools, their usefulness in clinical settings is limited by their low specificity and sensitivity, especially during the early stages of the disease (7).
Current treatment strategies for PDAC encompass surgical resection for localized cases, alongside chemotherapy and radiation therapy. The prevalent chemotherapy regimen for advanced PDAC is FOLFIRINOX, which consists of fluorouracil, leucovorin, irinotecan and oxaliplatin. This combination yields modest enhancements in OS relative to earlier treatment modalities (8). Furthermore, the pairing of gemcitabine and nab-paclitaxel is employed as a first-line approach, yielding moderate outcomes (9). Nevertheless, despite these therapeutic advancements, the prognosis for PDAC remains unfavorable, primarily due to its inherent resistance to chemotherapy and the presence of a dense desmoplastic stroma that obstructs effective drug delivery (10).
Given the aggressive nature of PDAC and the limited efficacy of current treatment options, there is an urgent need to discover new therapeutic targets. Molecular investigations of PDAC have identified several promising targets, such as KRAS mutations, which are present in ~90% of patients, along with heightened expression of epidermal growth factor receptor and vascular endothelial growth factor (11,12). However, clinical trials targeting these pathways have so far produced limited outcomes, underscoring the challenges in developing effective treatments for this malignancy (13–15).
To improve the outcomes of patients with PDAC, it is essential to develop more advanced prognostic models and tailored treatment strategies. The combination of molecular biomarkers, including genetic mutations, alongside clinical parameters, holds potential for forecasting patient outcomes and informing therapeutic choices (16). Furthermore, the advent of immunotherapy as a prospective treatment approach for PDAC, whilst still in its early phases, could provide renewed optimism for patients who have depleted standard treatment options (17). Immunotherapy approaches, especially those utilizing programmed cell death protein 1/programmed death-ligand 1 inhibitors, have demonstrated constrained efficacy in initial clinical trials. Nonetheless, the integration of immune checkpoint inhibitors with additional modalities like chemotherapy or targeted therapies could potentially improve the efficacy of these strategies (18).
In summary, PDAC presents a marked obstacle in the realm of cancer treatment, characterized by its rapid advancement, diagnosis at advanced stages and notable resistance to current therapeutic approaches. Despite advancements in therapeutic approaches, the outlook for patients with PDAC remains unfavorable. Continued research is essential for identifying novel biomarkers, uncovering new therapeutic targets and developing personalized treatment strategies that could improve clinical outcomes. The creation of new prognostic models incorporating genetic, molecular and clinical data is vital for advancing targeted therapies and enhancing survival rates for this devastating cancer.
Manganese (Mn) is a crucial trace element in biological systems, involved in regulating essential cellular functions such as antioxidant defense and enzyme activation. Mn serves as an important cofactor for several enzymes, particularly Mn-dependent superoxide dismutase (MnSOD), which is vital for maintaining cellular homeostasis. Whilst Mn is essential, excessive accumulation can lead to toxicity, particularly in the brain, where high Mn levels have been associated with neurodegenerative conditions such as Parkinson's disease and Huntington's disease (19,20). Beyond its neurotoxic effects, recent research indicates that Mn may also serve a role in the initiation and progression of cancer (21). However, its precise role in cancers, including PDAC, remains poorly understood.
Mn deficiency has been reported to induce cellular stress and activate pathways related to apoptosis and oxidative damage. A key mechanism by which Mn deficiency affects cellular function is through the activation of the tumor suppressor protein p53, which promotes mitochondrial-mediated apoptosis (22). Mn is crucial for maintaining the equilibrium of reactive oxygen species (ROS) within cells. Mn-dependent enzymes, particularly MnSOD, are vital components of the cellular antioxidant defense system. Imbalances in this system can lead to carcinogenesis. In cancer, Mn deficiency compromises the antioxidant defense, causing oxidative damage and genomic instability, which may lead to tumorigenesis (23).
Furthermore, research has reported that Mn modulates signaling pathways that support the survival, proliferation and spread of cancer cells. Specifically, in oral squamous cell carcinoma, Mn has been reported to activate the YAP/TAZ signaling pathway, enhancing ferroptosis, a type of cell death dependent on iron (24). Similarly, alterations in Mn metabolism have been associated with the progression of prostate cancer, with reduced serum Mn concentrations observed in patients. This suggests that Mn deficiency may contribute to cancer development (25).
Whilst marked advancements have been achieved in elucidating the function of Mn and Mn-related genes in cancer, their precise influence on the prognosis of PDAC is still inadequately investigated. The poor prognosis of PDAC is largely attributed to factors such as late-stage diagnosis, swift metastatic progression and a pronounced resistance to standard treatment modalities, including chemotherapy and radiation (26). Similar to other cancers, alterations in metal metabolism, particularly involving Mn, are thought to serve a role in the development and advancement of PDAC. Moreover, Mn is integral to cellular signaling, the management of oxidative stress and mitochondrial functionality, highlighting its potential as a focal point for further research in the realm of PDAC.
Recent studies have emphasized the significance of metabolism-related genes (MRGs) in cancer prognosis. In particular, Mn-associated pathways are thought to influence the tumor microenvironment (TME) and immune responses, offering potential insights for the development of new prognostic models for PDAC (27). However, the exact contributions of Mn-related genes to PDAC prognosis remain unclear. Therefore, the present study aimed assess the association between MRGs and PDAC outcomes through the evaluation of transcriptomic data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. Differential expression analysis was also performed to identify MRGs that are significantly altered in PDAC tissues. Furthermore, prognostic MRGs were identified through univariate Cox regression and Least Absolute Shrinkage and Selection Operator (LASSO) regression analyses, leading to the development of a predictive risk model. This model then underwent validation with independent PDAC cohorts. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed to provide insights into the biological pathways affected by these genes. Additionally, an evaluation of the immune microenvironment was performed to elucidate the influence of these MRGs on immune cell infiltration and their role in modulating immune responses in PDAC. Drug sensitivity analysis was performed to identify compounds targeting PDAC with disrupted Mn metabolism. Furthermore, reverse transcription-quantitative (RT-qPCR) and Kaplan-Meier (KM) survival analysis were used for experimental validation of key MRGs. This comprehensive methodology aimed to identify prognostic biomarkers, develop a risk model and investigate therapeutic strategies for improving the outcomes of patients with PDAC.
Materials and methods
Datasets
The TCGA-PDAC dataset was obtained from TCGA (https://portal.gdc.cancer.gov/), including 176 PDAC samples. Of these, 84 samples were from survivors and 92 from deceased patients, forming the training set. Additionally, the GSE62452 and GSE28735 datasets were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/) (28,29). These datasets were merged and served as the external validation set. After excluding samples lacking mRNA data, complete survival information and clinical pathological data, the final cohort included tissue samples from 107 patients with PDAC, consisting of 30 survivors and 77 deceased patients. The Genomics of Drug Sensitivity in Cancer database (https://www.cancerRxgene.org/) was used to obtain IC50 values for 198 drugs, which were utilized to assess drug sensitivity in PDAC samples.
Mn MRGs as the target gene set
MRGs were defined as genes associated with Mn metabolism. Initially, 1,829 genes were identified from the GeneCards database (https://www.genecards.org/) using ‘Mn metabolism’ as the search keyword (Table SI). To confirm their functional relevance, GO and KEGG analyses were performed.
Gene set enrichment analysis (GSEA) of Mn metabolism pathways
A predetermined MRG set (derived from the GeneCards database using ‘Mn metabolism’ as the search key word) was used to perform GSEA to further elucidate the function of Mn metabolism in PDAC. Expression data (sourced from TCGA-PDAC and GEO datasets: GSE62452 and GSE28735) collected from PDAC samples were subjected to the GSEA. GSEA was run once for the whole collection of genes, a second time for genes demonstrating reduced expression (in PDAC tumor tissues compared to normal controls, defined by log2FC <-1.5 and adj.P<0.05) and a third time for genes with higher expression (in PDAC tumor tissues compared to normal controls, defined by log2FC >1.5 and adj.P<0.05).
Gene Set Variation Analysis (GSVA)
GSVA was performed utilizing the GSVA package (version 2.2.0; Bioconductor, http://www.bioconductor.org/packages/release/bioc/html/GSVA.html) to assess variations in signaling pathways among the high- and low-risk groups. The analysis was conducted using a significance threshold defined by an adjusted P-value of <0.05. The GSVA algorithm was applied to evaluate the enrichment of predefined gene sets, with the x-axis representing the t-value calculated using a t-test. Larger t-values indicate more significant differences between groups, with blue bars representing upregulated pathways and green bars representing downregulated pathways.
Identification of differentially expressed MRGs
Under the conditions of |log2FoldChange (FC)| >1.5 and adj.P<0.05, the DESeq2 program (version 1.30.1; Bioconductor, http://bioconductor.org/packages/DESeq2) was used to identify differentially expressed genes (DEGs) between the PDAC and control groups. Subsequently, the biological roles of the DEGs were assessed by performing analyses in GO and KEGG using the ‘clusterProfiler’ software (version 4.0.5; Bioconductor, https://bioconductor.org/packages/clusterProfiler; adj.P<0.05). By intersecting the DEGs with Mn MRGs, the ‘VennDiagram’ tool in R (version 1.7.3; CRAN, http://cran.r-project.org/package=VennDiagram) was utilized to elucidate possible candidate genes. A Venn diagram was then used to identify the overlap. The candidate genes were subjected to additional GO and KEGG analyses using the ‘clusterProfiler’ package, and the ‘GOplot’ program (version 1.0.2; CRAN, http://cran.r-project.org/package=GOplot) was used to display the top five enriched pathways.
Tumor Immune Dysfunction and Exclusion (TIDE) Score Analysis
The TIDE score was calculated using the TIDE algorithm (version 1.0.0, http://github.com/nangong/TIDE) to assess tumor immune dysfunction and exclusion in both high- and low-risk groups. The TIDE algorithm integrates tumor expression profiles with single-cell reference datasets to infer the degree of immune dysfunction and exclusion within the tumor microenvironment. The TIDE score is composed of two components: The dysfunction score, which reflects the impairment of T cell function, and the exclusion score, which indicates the degree to which T cells are excluded from the tumor. A higher TIDE score is associated with poorer clinical outcomes.
Regulatory and co-expression network analysis
To investigate molecular regulatory mechanisms, miRNAs associated with the prognostic genes were predicted using the miRWalk database (version 2.0, http://mirwalk.umm.uni-heidelberg.de/) and miRNet (version 2.0, http://www.mirnet.ca/). miRNAs identified by both databases were selected as key candidates. Potential lncRNAs interacting with these miRNAs were predicted using StarBase (version 3.0, http://starbase.sysu.edu.cn/) and miRNet (version 2.0). The intersecting lncRNAs from both databases were chosen as key lncRNAs. The lncRNA-miRNA-mRNA regulatory network was visualized using Cytoscape (version 3.9.1; http://cytoscape.org/). Additionally, the GeneMANIA database (version 3.6.0, http://genemania.org/) was used to identify genes functionally related to the prognostic genes and construct a co-expression network.
Development and validation of the MRGs prognostic model
Gene selection was performed using univariate Cox regression, multivariate Cox models and LASSO regression to elucidate genes associated with PDAC prognosis. Using the survival program (version 3.2–13; CRAN, http://cran.r-project.org/package=survival), PDAC samples from the training cohort with full survival data were first subjected to univariate Cox regression. The identification of genes with a strong association with PDAC survival was performed using the criterion of hazard ratio (HR) ≠1 and P<0.01. Further refinement of the results was performed using the proportional hazards (PH) assumption test, with a significance threshold of P>0.05. Genes that passed the PH assumption test (P>0.05) and the univariate Cox regression criteria (P<0.01) were then analyzed using the glmnet software (version 4.1–3; CRAN, http://cran.r-project.org/package=glmnet) for LASSO regression. To optimize gene selection, L1 regularization was first used to minimize the gene coefficients, and then 10-fold cross-validation was performed. A total of 12 genes were used to build the risk model after they were identified to be significant prognostic factors. The risk score for every PDAC sample was determined using the following algorithm, taking into account all available survival data from the training and validation cohorts: Risk score = ∑ni=1 (βi × xi). In this model: βi, coefficient of each prognostic gene; i, multivariate Cox regression model; xi, expression level of prognostic gene, i. The PDAC samples that had available survival data were divided into two groups: High-risk and low-risk, according to the median risk score. These groups were assessed for clustering using principal component analysis (PCA). The survival package was utilized to perform a Kaplan-Meier survival analysis, and a log-rank test was used to determine whether there were statistically significant differences between the survival curves of the two groups. Using ggplot2 software (version 3.3.5; CRAN, http://cran.r-project.org/package=ggplot2), Receiver Operating Characteristic (ROC) curves were created to assess the predictive performance of the model across a 1–5 year time frame. The area under the curve was computed as part of the performance evaluation using the timeROC program (version 0.4; CRAN, http://cran.r-project.org/package=timeROC). For improved visualization of survival distributions in both risk groups, survival curves and status plots were generated using the Survminer package (version 0.4.9; CRAN, http://cran.r-project.org/package=survminer). Furthermore, to demonstrate how the expression patterns of the prognostic genes varied among different groups, a heatmap was generated using the ggheat function in the tinyarray package (version 2024; GitHub, http://github.com/xjsun1221/tinyarray). To ensure the robustness of the model, it was externally validated using the GSE62452 and GSE28735 datasets. These external datasets were subjected to PCA once more to assess clustering. ROC curves were then utilized to evaluate the performance of the models and Kaplan-Meier survival analysis and log-rank tests were performed to validate them on the test set. Additionally, heatmaps of the prognostic gene expression patterns, survival status plots and risk curves were generated for the test set.
To assess the association between clinical pathological factors and the prognostic model, the survival package was used to perform univariate and multivariate Cox regression analyses on PDAC samples with full survival data from the training set. Clinical variables, such as age and sex were analyzed, as well as pathological stages [metastasis (M), lymph node (N) and tumor (T) stages, and tumor stage] and the risk score. In the multivariate analysis, factors were considered to be independent prognostic markers if they met the PH assumption (P>0.05).
Furthermore, the rms program was used to create a nomogram that predicted the survival probabilities for patients with PDAC at 1, 3 and 5 years using the independent prognostic factors revealed in the training set. The prediction accuracy of the nomogram was evaluated by creating ROC curves for time periods ranging from 1–5 years using the riskRegression software. The performance of the nomogram was compared with those of other independent prognostic indicators using these curves. To assess specificity and sensitivity, the AUC was computed. In addition, the predictive accuracy of the nomogram was evaluated by drawing calibration curves for 1–5 years and comparing the anticipated survival probability with theoretical survival curves. By comparing the net benefit of the nomogram model with that of other independent predictive indicators, the rmda program was used to perform a decision curve analysis (DCA) evaluation of the clinical utility of the nomogram.
Clinical feature association analysis
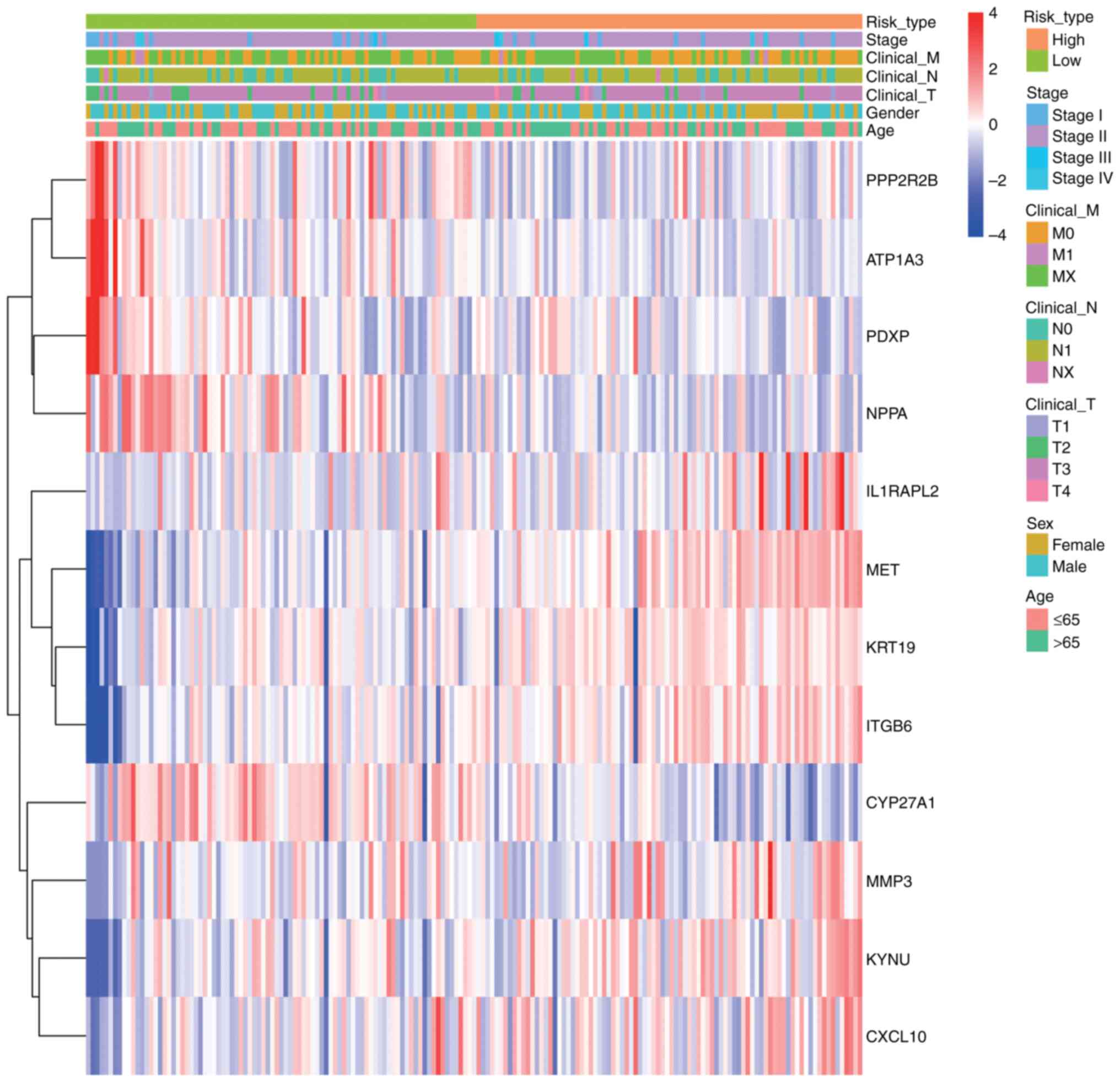
Using the pheatmap software, a heatmap was created to assess the relationships between the risk score, prognostic genes and clinical features in PDAC samples and the whole survival data of the training cohort. Using risk ratings and several clinical variables, this heatmap demonstrated how the predictive genes were expressed. The Wilcoxon rank-sum test or χ2 test (P<0.05) was used to evaluate differences in clinical features between the two groups, and the findings were displayed in a heatmap. To further distinguish between the high- and low-risk groups, functional enrichment analysis was used to find DEGs.
Cell culture
The human PDAC cell lines PANC-1 (RRID: CVCL_0480) and PaTu-8988t (RRID: CVCL_1847) were purchased from the Cell Bank of the Chinese Academy of Sciences, and the human pancreatic duct epithelial cell line hTERT-HPNE (RRID: CVCL_C466) was purchased from the American Type Culture Collection (ATCC). All cell lines were authenticated using short tandem repeat profiling and routinely tested for mycoplasma contamination using Mycoplasma Removal Agent (cat. no. BL591B; Biosharp Life Sciences).
PANC-1 and PaTu-8988t cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; cat. no. MA0212; MeilunBio®; Dalian Meilun Biotech Co., Ltd.) supplemented with 10% fetal bovine serum (FBS; cat. no. S1001-500; BIOAGRIO; Shanghai Yuli Biotechnology Co., Ltd.) and 1% penicillin-streptomycin (cat. no. C100C5; New Cell and Molecular Biotech Co., Ltd.). They were maintained in a humidified incubator (Heal Force Bio-meditech Holdings Ltd.) at 37°C with 5% CO2.
hTERT-HPNE cells were cultured according to ATCC recommendations using a base medium composed of 75% glucose-free DMEM (cat. no. D5030; Sigma-Aldrich; Merck KGaA) and 25% M3:BaseF™ medium (cat. no. M300F-500; INCELL Corporation LLC), supplemented with 5% FBS (FBS; cat. no. S1001-500; BIOAGRIO; Shanghai Yuli Biotechnology Co., Ltd.), 10 ng/ml recombinant human EGF (PeproTech China), 5.5 mM D-glucose and 750 ng/ml puromycin (Sigma-Aldrich; Merck KGaA). Cells were maintained in T25/T75 culture flasks with medium refreshed every 2–3 days and subcultured at 80–90% confluence using 0.25% trypsin-EDTA.
Transfection
Kynureninase (KYNU) gene knockdown was performed in human PDAC PANC-1 (RRID: CVCL_0480) and PaTu-8988t (RRID: CVCL_1847) cell lines using small interfering (si)RNAs synthesized by Bioscien Biotech Co., Ltd. The sequences used were as follows: KYNU-siRNA, 5′-CCTCCAGTTGATTTATCATTAtt-3′ and negative control (NC)-siRNA, 5′-UUCUCCGAACGUGUCACGUtt-3′. siRNAs (6 pmol/well) were diluted in serum-free Opti-MEM™ (Gibco; Thermo Fisher Scientific, Inc.) and mixed with Lipofectamine™ RNAiMAX Transfection Reagent (cat. no. 13778075; Invitrogen™; Thermo Fisher Scientific, Inc.). After 15 min of incubation at room temperature, the mixture was added to the cells. Cells were incubated at 37°C for 6 h, after which the medium was replaced with complete growth medium. Cells were harvested at 48 h post-transfection for downstream analysis.
For Cytochrome P450 Family 27 Subfamily A Member 1 (CYP27A1) overexpression, pcDNA3.1-CYP27A1 and the corresponding empty vector were purchased from Shanghai GenePharma Co., Ltd. Transfection was performed using Lipofectamine 3000 (cat. no. L3000015; Invitrogen; Thermo Fisher Scientific, Inc.), with 2 µg plasmid DNA/well, following the manufacturer's instructions. Cells were collected for analysis at 48 h after transfection. All experiments were independently performed in triplicate to ensure reproducibility.
RT-qPCR validation
Total RNA was extracted from PDAC cell lines (PANC-1 and PaTu-8988t) using the RNA extraction kit (cat. no. AC0202; Shandong Sikejie Biotechnology Co., Ltd.). A 20 µl reverse transcription reaction was prepared for cDNA synthesis, utilizing the ABScript Neo RT Master Mix for qPCR with gDNA Remover (cat. no. RK20433; ABclonal Biotech Co., Ltd.). Reverse transcription was performed as follows: 37°C for 2 min (pre-denaturation), 55°C for 15 min (cDNA synthesis) and 85°C for 5 min (enzyme inactivation). qPCR was performed using 10 µl 2X Universal SYBR Green Fast qPCR Mix (cat. no. RK21203; ABclonal Biotech Co., Ltd.). The thermocycling protocol included an initial denaturation at 95°C for 3 min, followed by 40 cycles of denaturation at 95° for 5 sec and annealing at 60°C for 30–34 sec. Table SII presents the primer sequences. The 2−ΔΔCq method was used to calculate relative mRNA levels, which were then normalized to the control group (30). Sangon Biotech Co., Ltd. synthesized the primers.
Cell viability assay
A Cell Counting Kit-8 (CCK-8; cat. no. MA0218-1; MeilunBio; Dalian Meilun Biotech Co., Ltd.) was used to measure cell viability in human PDAC PANC-1 (RRID: CVCL_0480) and PaTu-8988t (RRID: CVCL_1847) cell lines. Briefly, cells in the logarithmic phase of growth were treated with 0.25% trypsin, suspended in medium supplemented with 10% FBS and plated in 96-well plates at a density of 5,000 cells/well. After the cells adhered, the culture medium was replaced with 10 µl CCK-8 solution and 90 µl fresh medium, and the cells were incubated for 2 h. The absorbance was then measured at 450 nm.
Colony formation assay
Pancreatic cancer 8988 and PANC-1 cell lines from both experimental and control groups were harvested during the logarithmic growth phase. Cell counting was performed using an automated cell counter (Countstar Rigel S2, Countstar, Inc.). Subsequently, 800 viable cells per well were seeded in 6-well plates for further experiments. Following seeding, the cells were maintained at 37°C under 5% CO2 with 95% relative humidity for 14 days, with medium changes performed every 48 h until each cellular clone contained >50 cells. Thereafter, cells were fixed with 4% paraformaldehyde at room temperature (20–25°C) for 20 min and stained with 0.1% crystal violet at room temperature for 30 min. Images were then captured and analyzed using lmageJ software (National Institutes of Health).
Wound healing assay (WHA)
The WHA was initiated by seeding 8988 and PANC-1 cell lines in 6-well plates to achieve ~90% confluence. Following 24-h of serum starvation, a sterile 200 µl pipette tip was used to scratch the cells in each well. Wound closure was observed under a microscope (Nikon Eclipse Ts2; Nikon Corporation) and images were captured at 0, 24 and 48 h post-scratch. The wound area was analyzed using ImageJ software (National Institutes of Health) and the migration rate was calculated using the following equation: Migration Rate=1-(Wound Area at tn/Wound Area at t0), where t0 is the initial wound area, tn is the wound area after n hours of the initial scratch.
Statistical analysis
The DESeq2 program was used to identify DEGs, with a cutoff of |log2FC| >1.5 and an adjusted P-value of <0.05. To evaluate OS among different patient groupings, log-rank tests and Kaplan-Meier survival curves were utilized. Spearman's rank correlation analysis was performed to assess the correlation between immune cell infiltration levels and the expression of prognostic genes. To identify the independent prognostic significance of risk factors, univariate and multivariate Cox regression models were used. Furthermore, genes that were revealed to have predictive value for PDAC were identified using a LASSO regression model. Using the Mann-Whitney U test, the high- and low-risk groups were utilized to assess differences in immune cell infiltration and activation of immunological pathways. For expression analysis, RT-qPCR data were analyzed using two-tailed unpaired Student's t-tests (each sample was analyzed in triplicate independent experiments), and results are presented as mean±standard error of the mean. Furthermore, for functional validation assays, comparisons between two groups were performed using two-tailed unpaired Student's t-tests (each experiment was repeated three times independently). R (version 4.0.2; The R Foundation) was used to perform all statistical analyses. P<0.05 was considered to indicate a statistically significant difference. The analytical framework of the present study is presented in Fig. 1.
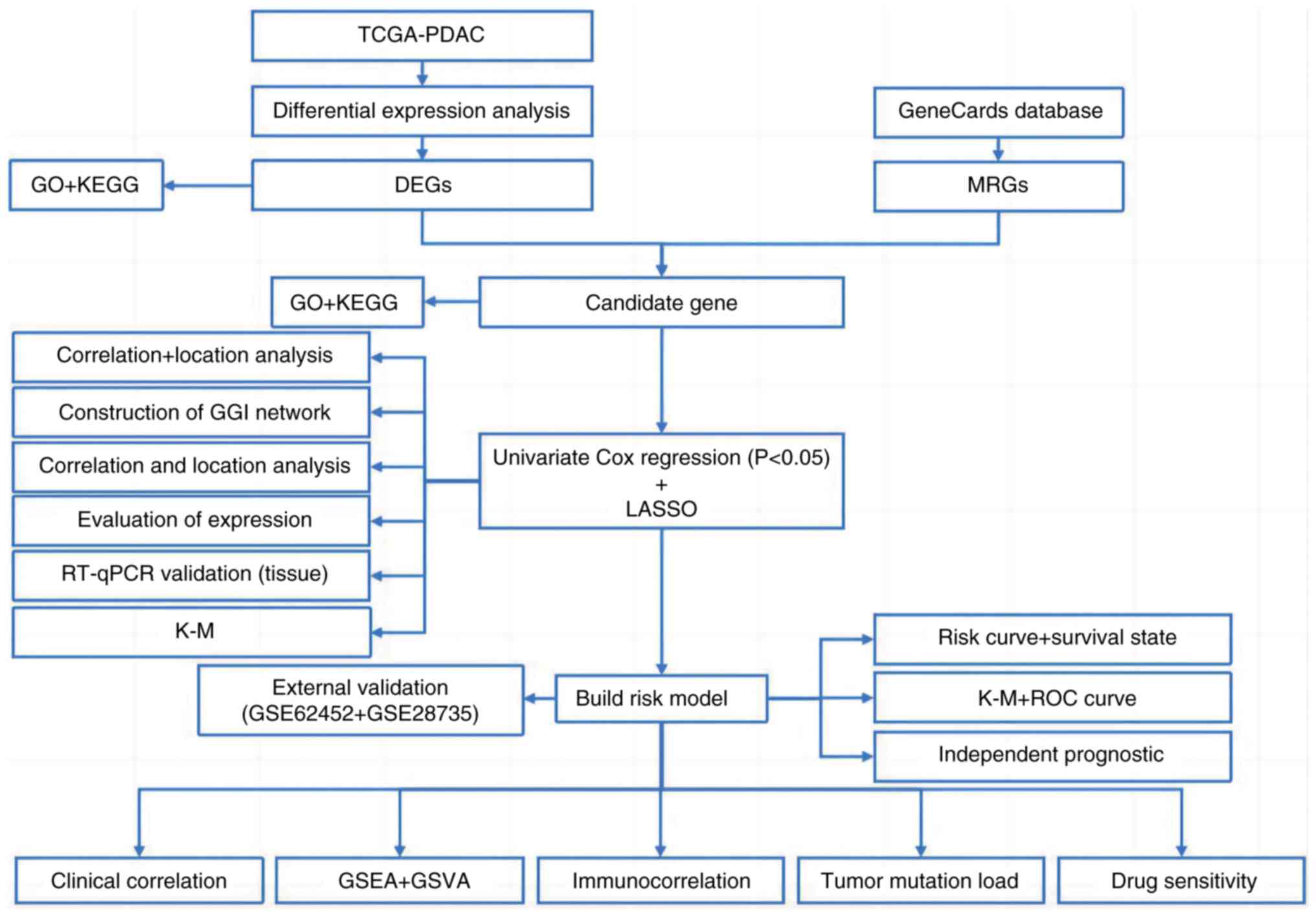
Results
DEGs between normal and tumor tissues
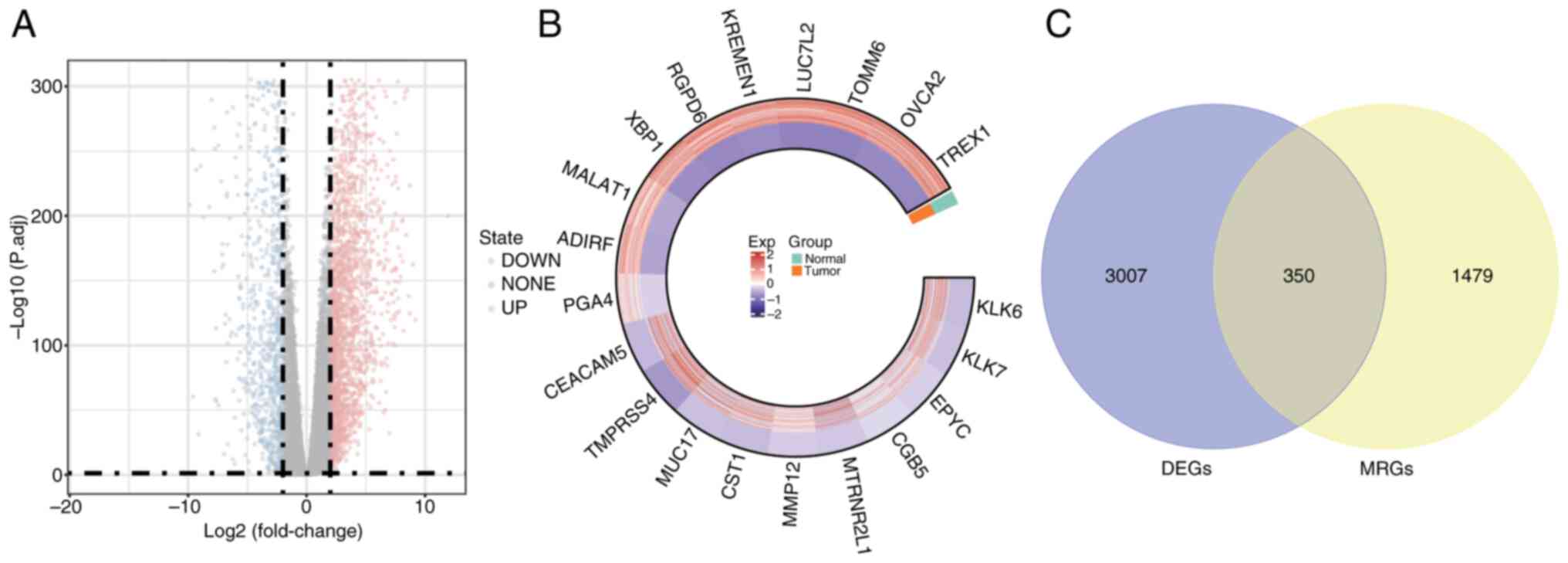
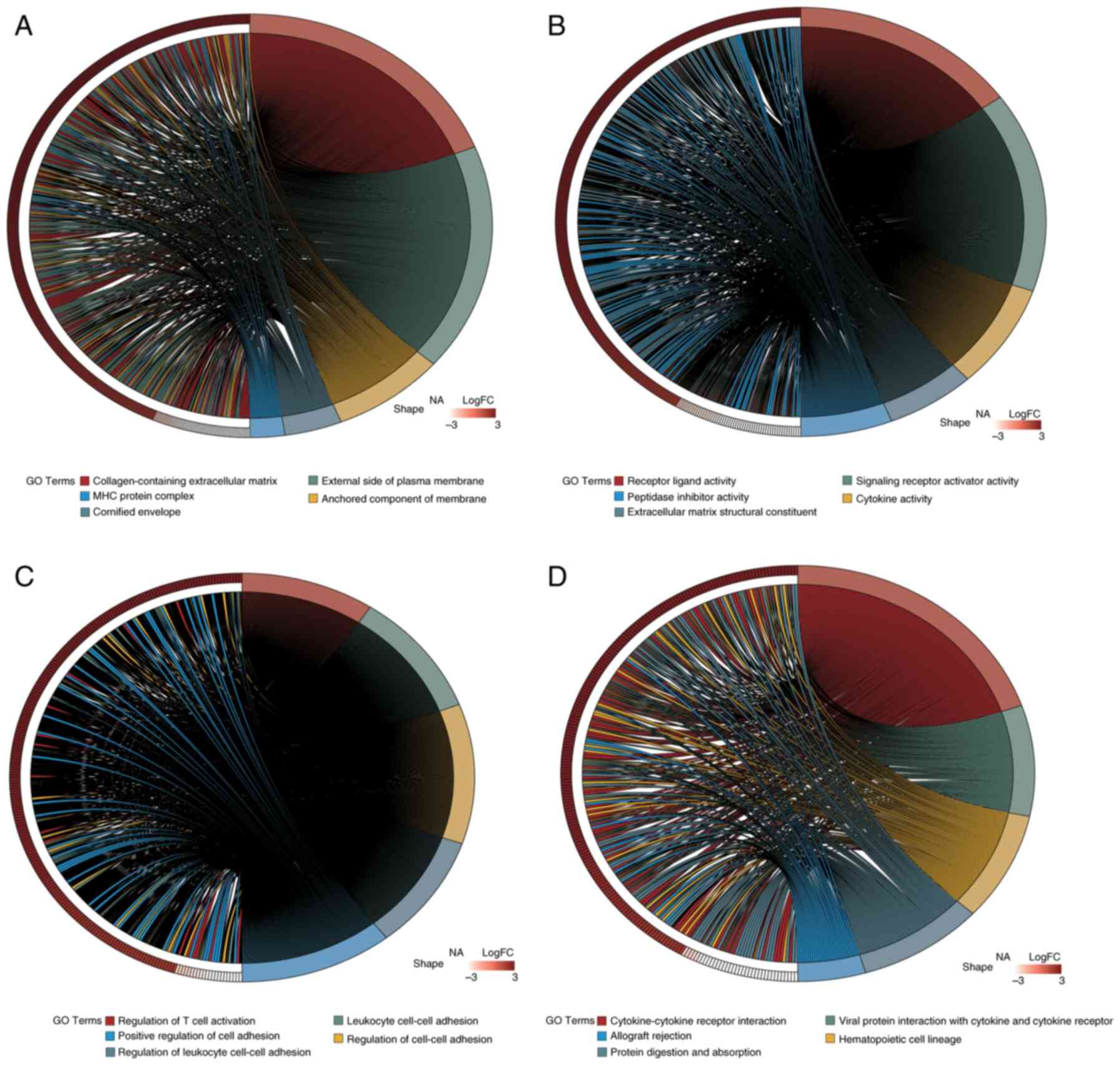
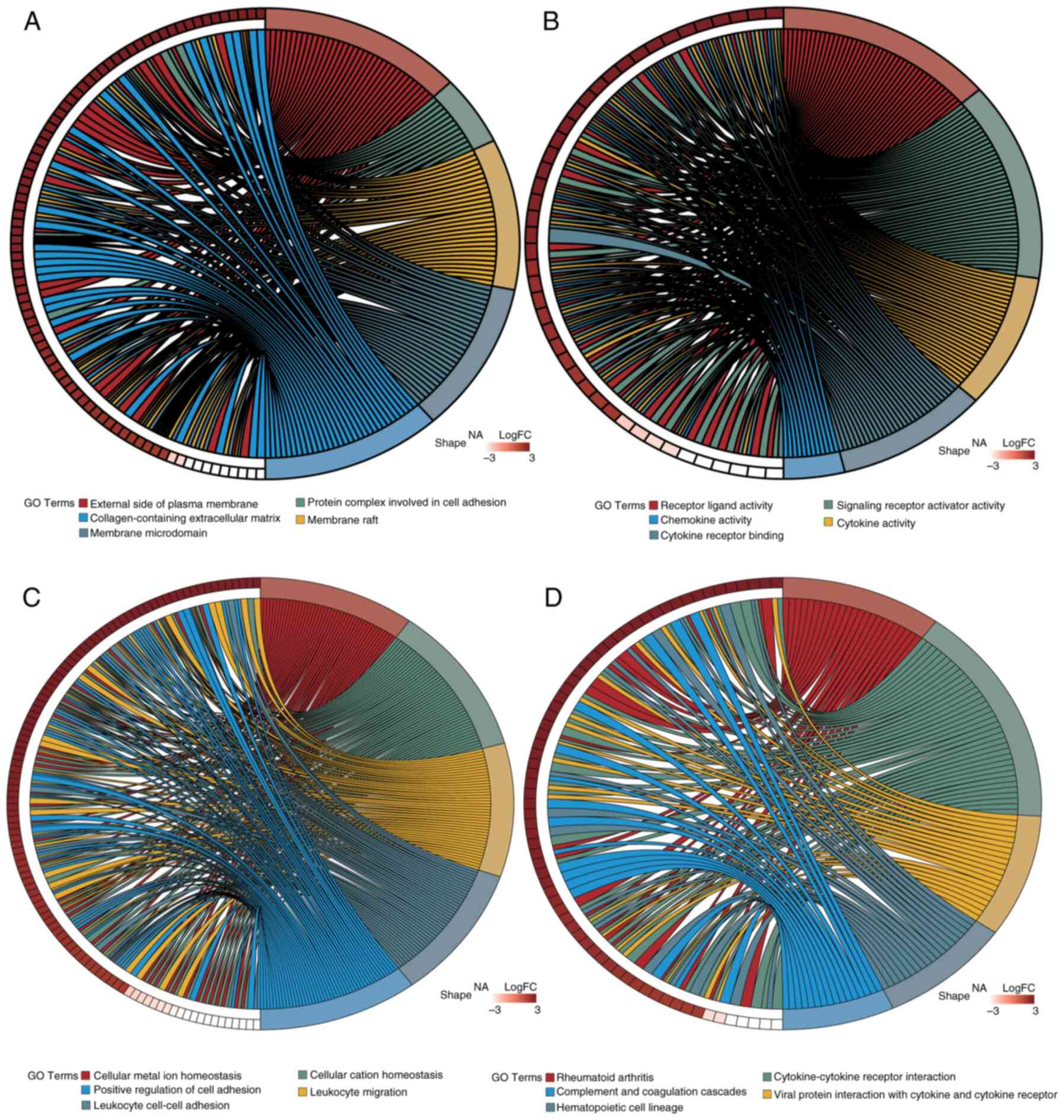
The DESeq2 package was utilized for the differential expression analysis of PDAC tumor samples compared with normal control tissues. The following criteria were used: |log2FC| >1.5 and adj.P<0.05. Table SIII presents the 3,357 DEGs identified in PDAC samples relative to normal controls: 2,264 of these genes were upregulated and 1,093 were downregulated. To visualize the distribution of these DEGs, a volcano plot was generated using the ‘ggplot2’ R package, highlighting the top 10 genes that were upregulated and downregulated (Fig. 2A). Additionally, the expression of 20 DEGs was visualized in a circos plot generated using the ‘circlize’ R package (Fig. 2B). GO analysis (Fig. 3A-C) and KEGG pathway enrichment analysis (Fig. 3D) were performed using the clusterProfiler package. The DEGs were categorized into Biological Process (BP), Cellular Component (CC) and Molecular Function (MF) groups. Utilizing the ‘GOplot’ R package, the top five enriched pathways were represented. BP pathways encompassed the regulation of T cell activation, leukocyte cell-cell adhesion and positive regulation of cell adhesion; MF pathways included receptor-ligand activity and cytokine activity; and CC pathways were associated with collagen-containing extracellular matrix and major histocompatibility complex protein complexes. Significantly enriched KEGG pathways (adj.P<0.05) included interactions between cytokines and their receptors, the lineage of hematopoietic cells and the digestion and absorption of proteins were all KEGG pathways.
Functional enrichment analysis
To assess the relevance of 1,829 MRGs, GO and KEGG analyses were performed. The GO analysis (Table SIV) identified significant enrichment in processes such as Mn ion binding, metal ion transport and oxidation-reduction. Additionally, the KEGG analysis (Table SV) demonstrated significant associations with pathways related to metal ion metabolism, underscoring the functional categorization of the gene set in the present study in relation to Mn metabolism.
GSEA for Mn metabolism pathways in PDAC
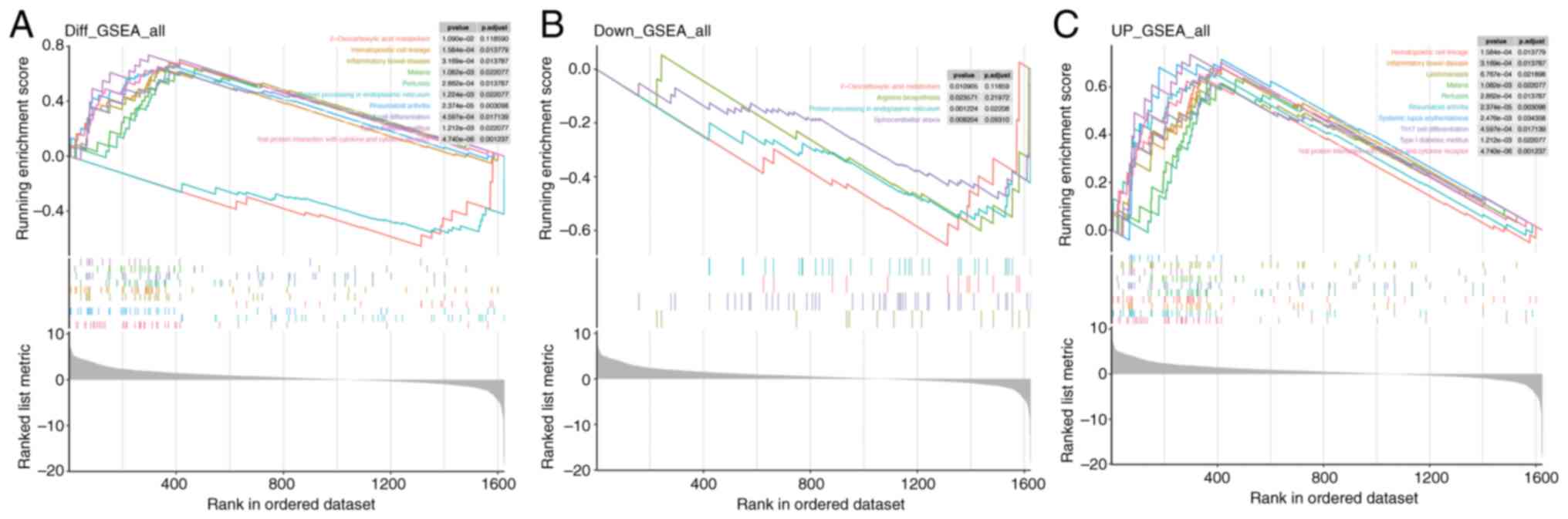
In the overall GSEA results for Mn metabolism pathways in PDAC (Fig. 4A), pathways such as ‘2-Oxocarboxylic acid metabolism’ and ‘Hematopoietic cell lineage’ were significantly enriched. For the downregulated genes (Fig. 4B), enrichment was noted in ‘2-Oxocarboxylic acid metabolism’, ‘Arginine biosynthesis’, ‘Protein processing in endoplasmic reticulum’ and ‘Spinocerebellar ataxia.’ Conversely, the upregulated gene set (Fig. 4C) showed enrichment in ‘Hematopoietic cell lineage’. These results indicate that Mn metabolism-related genes in PDAC are implicated in a wide range of BPs, spanning metabolic and immune/inflammatory pathways.
Identification and enrichment analysis of candidate genes
To identify potential candidate genes, the ‘VennDiagram’ package in R was utilized to intersect DEGs with MRGs, resulting in a Venn diagram. A total of 350 genes (accounting for 7.24% of the total) were identified and selected for further examination (Fig. 2C). Furthermore, to evaluate the functional roles of these candidate genes and their involvement in several biological processes, enrichment analyses for GO and KEGG were performed using the ‘clusterProfiler’ R package. A total of 1,721 GO terms and 80 KEGG pathways met the significance threshold of adj.P<0.05. Of these, 1,529 related to BP, 61 to CC and 131 to MF. The most enriched GO and KEGG pathways were visualized using the ‘GOplot’ R package (Fig. 5). Enriched BP terms included ‘cellular metal ion homeostasis’, ‘leukocyte migration’ and ‘positive regulation of cell adhesion’. MF terms included ‘receptor-ligand activity’, ‘cytokine activity’ and ‘chemokine activity’. CC terms included ‘protein complexes in cell adhesion’ and ‘collagen-containing extracellular matrix’. Key KEGG pathways included ‘Rheumatoid arthritis’, ‘Cytokine-cytokine receptor interaction’ and ‘Complement and coagulation cascades’.
Development of a prognostic gene model in TCGA cohort
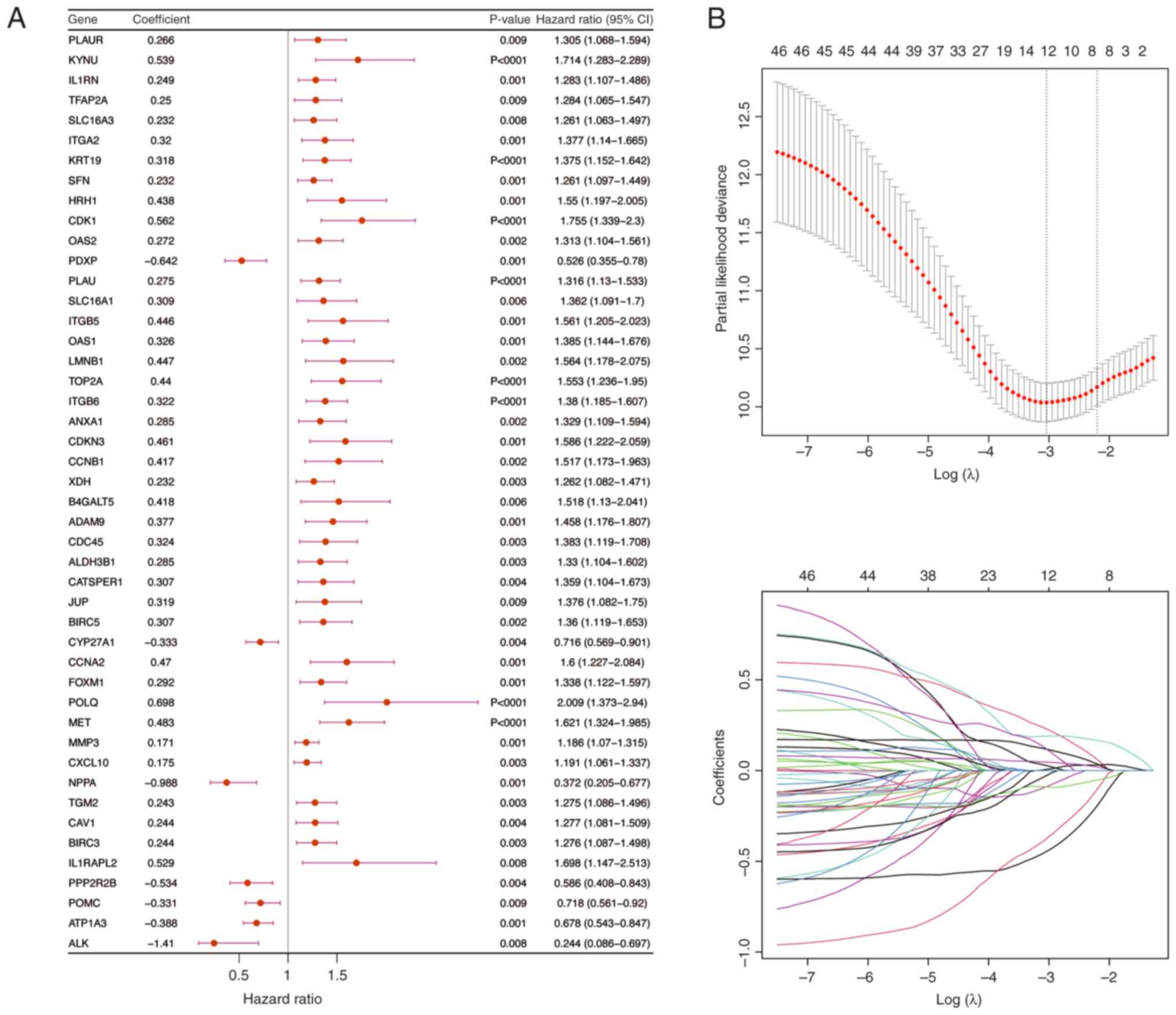
Univariate Cox regression analysis was performed using PDAC samples that included full survival data from the training cohort (Fig. 5A). The results identified 46 genes that were associated with survival. Subsequently, the PH assumption test was used to assess these genes (P>0.05) and then a 10-fold cross-validation procedure was used to select genes for LASSO regression analysis if P<0.01 in the univariate Cox analysis. The final set of 12 prognostic genes was identified (Fig. 6B) and then a risk score was computed for every PDAC sample in the validation and training cohorts using these genes.
The samples were divided into two categories, high-risk and low-risk, according to their median risk scores. To compare the distribution of the high-risk and low-risk groups, PCA was used (Fig. 7A). The survival package was used to perform a Kaplan-Meier survival analysis, which demonstrated that the high-risk group had significantly worse survival outcomes compared with the low-risk group (Fig. 7B; P<0.05).
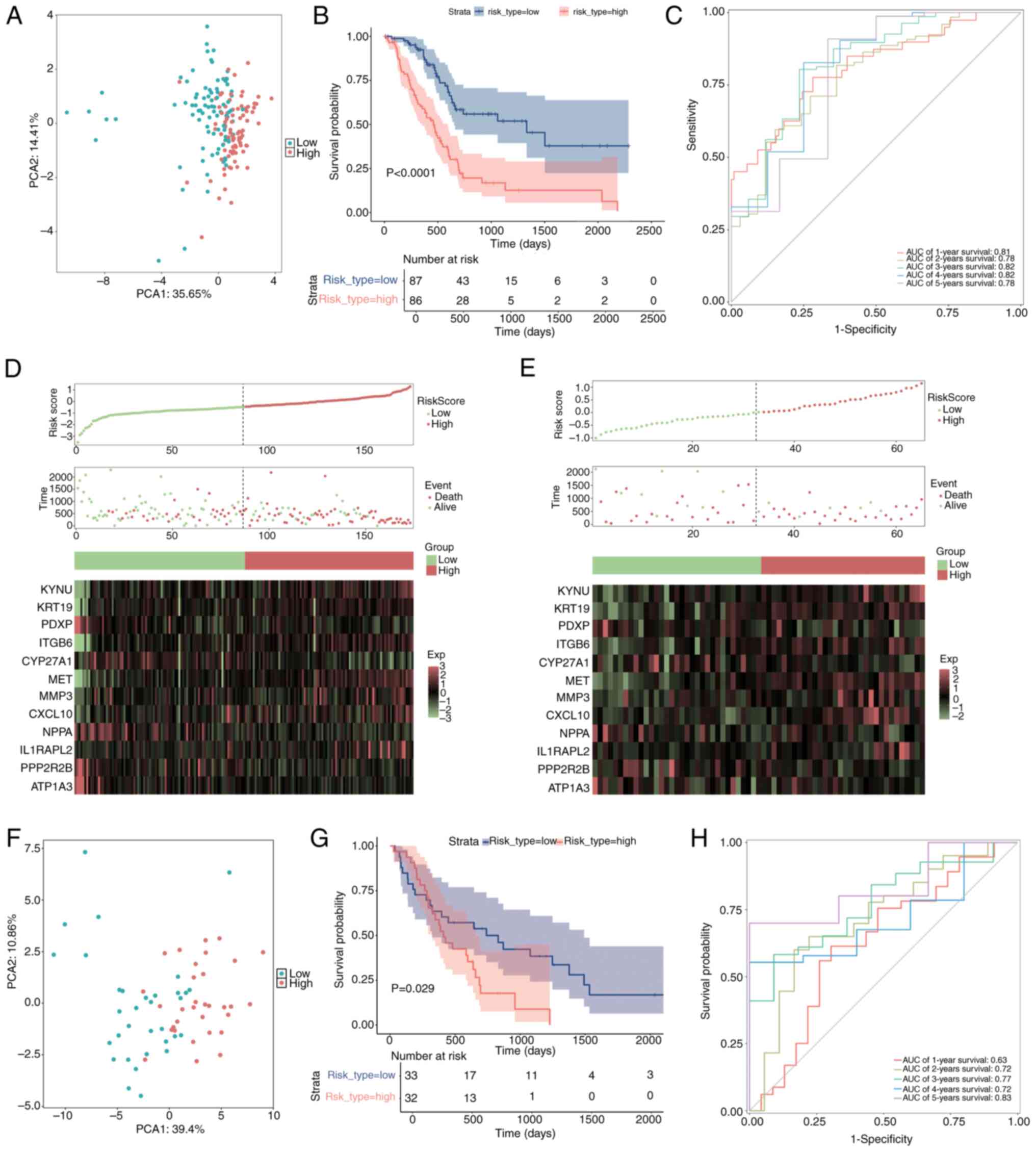
Using ggplot2-generated ROC curves, survival rates were then assessed from 1–5 years. AUC was computed using the timeROC package to evaluate the prediction ability of the model (Fig. 7C). For every time point, the AUC values were >0.7, demonstrating that the model was relatively accurate. Moreover, to further elucidate the distributions of survival in both risk groups, Survminer software was used to construct survival curves and status plots. Additionally, a heatmap illustrating the expression levels of the prognostic genes in these groups was created using the ggheat function from the tinyarray package (Fig. 7D).
External validation of the risk signature
To assess the broader applicability of the risk model, external validation was performed utilizing a merged dataset from GSE62452 and GSE28735 (Fig. 7E-H). The aggregation of the testing cohort as determined by PCA is illustrated in Fig. 7F. Kaplan-Meier survival analysis was performed utilizing the survival package, alongside log-rank tests, to assess and compare the survival outcomes of the high- and low-risk groups (Fig. 7G; P<0.05). The findings indicated a notable disparity, as the high-risk cohort exhibited a less favorable outcome compared with the low-risk cohort. To evaluate the precision of the predictions of the model within the validation cohort, a ROC curve analysis was performed (Fig. 7H). The AUC values across all time points were >0.6, suggesting a satisfactory level of predictive accuracy for the model. Additionally, survival status plots and risk curves were created to illustrate the survival distributions within the two risk categories in the validation cohort. A heatmap was generated to illustrate the expression patterns of prognostic genes concerning the two risk groups (Fig. 7E).
Independent prognostic significance of the risk model
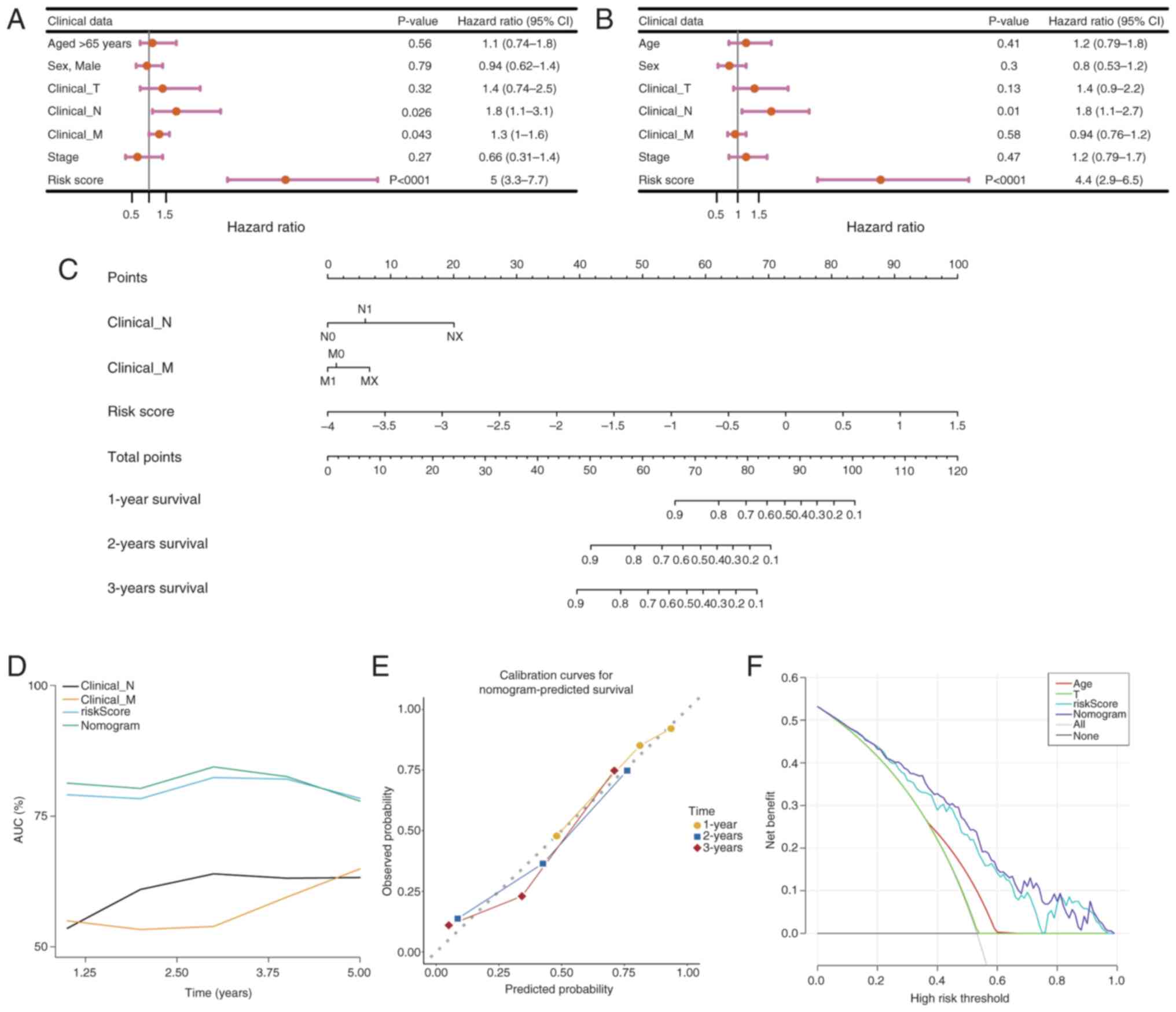
To assess the association between clinicopathological factors and the prognostic risk model, both univariate and multivariate Cox regression analyses were performed on PDAC samples that had comprehensive survival data. Clinical variables including age, sex and the pathological stages M, N and T, as well as tumor stage, were methodically integrated into the analysis utilizing the survival package. Variables that demonstrated significance in the univariate analysis (P<0.05) underwent additional scrutiny using the PH assumption test (P>0.05) prior to their incorporation into the multivariate model. The evaluation revealed that an age of >65 years, T stage and risk score served as independent prognostic indicators for PDAC (Fig. 8A and B). Subsequently, a nomogram was developed utilizing these independent prognostic factors to estimate the survival probabilities at 1, 3 and 5 years for patients with PDAC, employing the rms package (Fig. 8C). The performance of the nomogram was evaluated using ROC curves for survival predictions at 1-, 3- and 5-year intervals, comparing the accuracy of the model with other independent prognostic factors using the riskRegression package (Fig. 8D). The AUC was calculated to determine the sensitivity and specificity of the model in predicting survival outcomes. AUC values of ~1 indicate excellent predictive accuracy, whilst values of >0.6 denote robust model performance. Moreover, calibration curves for 1–5 year survival were constructed to evaluate the predictive accuracy of the nomogram by juxtaposing the predicted values with the ideal theoretical curve (Fig. 8E). A slope of ~1 signifies an ideal performance of the model. Furthermore, a DCA was performed utilizing the rmda package (Fig. 8F) to assess the clinical utility of the nomogram, quantifying the net benefit in comparison with other independent prognostic factors. A net benefit of >0 indicates the considerable importance of the model in the realm of clinical decision-making.
Clinical feature correlation analysis
To evaluate the associations among risk scores, prognostic genes and clinical characteristics in PDAC samples, a heatmap was constructed utilizing the pheatmap package. This illustrates the expression patterns of prognostic genes in conjunction with clinical features and associated risk scores, with the variations in clinical variables between the two groups statistically evaluated (P<0.05; Fig. 9). The heatmap revealed distinct clustering patterns between high- and low-risk groups. Key clinical features, including advanced tumor stage (T3-T4), lymph node metastasis (N1) and distant metastasis (M1), were significantly enriched in the high-risk group (Wilcoxon test, P<0.05).
GSEA and Gene Set Variation Analysis (GSVA)
The clusterProfiler package was utilized to perform GSEA, aiming to identify signaling pathways that are significantly enriched between the high- and low-risk groups, as determined by DEGs. Pathways exhibiting a |Normalized Enrichment Score (NES)| of >1 and an adjusted P-value of <0.05 were deemed significantly enriched (Fig. 10A and B) In Fig. 10A, the GO enrichment analysis results are displayed. The horizontal axis represents the enrichment score ranging from −4 to 4, reflecting the degree of gene enrichment or depletion in specific biological processes. The vertical axis lists various biological processes related to skin development, keratinization and epithelial cell differentiation, among others. The color of the curves indicates the adjusted p-value, with red signifying higher significance (1×10−4) and blue indicating lower significance (2×10−4). Notably, processes such as ‘Epidermis development’ and ‘Keratinization’ show prominent peaks, suggesting their significant enrichment in the DEGs. Similarly, Fig. 10B presents the KEGG pathway enrichment analysis. It follows the same format as Fig. 10A, with the vertical axis showing different KEGG pathways associated with tumors, cellular responses and carcinogenesis. Prominent peaks in pathways like ‘Tumor differentiated well vs. poorly dn’ and ‘Breast cancer basal up’ highlight their significant enrichment in the DEGs. These findings indicate that the enriched biological processes and pathways may play crucial roles in distinguishing the high- and low-risk groups, providing insights into the molecular mechanisms underlying the observed differences. Furthermore, GSVA was performed utilizing the GSVA package to assess variations in signaling pathways among the groups, employing a significance threshold defined by an adjusted P-value of <0.05 (Fig. 10C). The results of GSVA are displayed in Fig. 10C. The horizontal axis denotes the enrichment score, and the vertical axis lists various signaling pathways. The color of the curves indicates the degree of pathway variation among the groups.
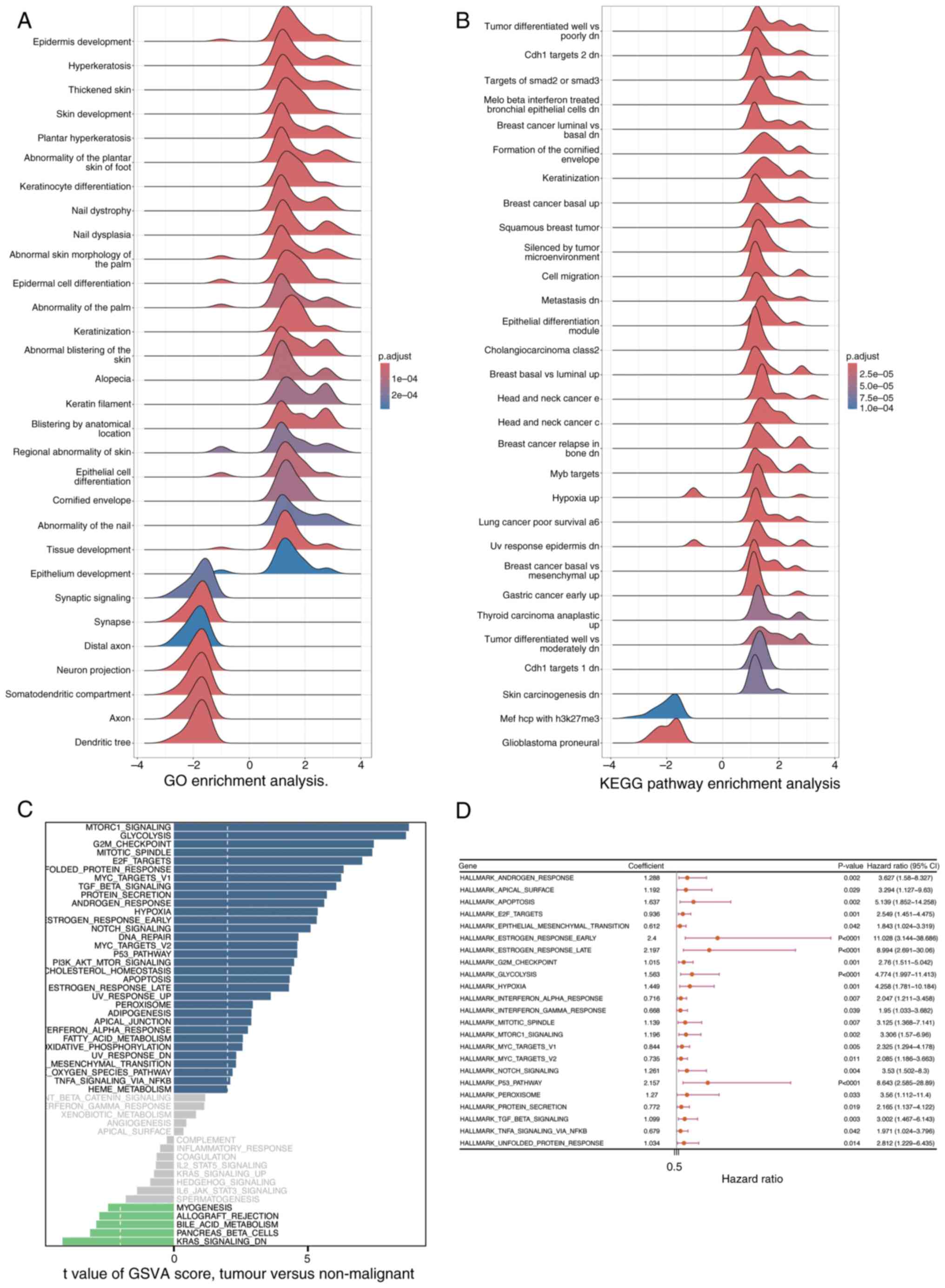
Subsequently, Univariate Cox regression analysis was performed using the survival package to assess the association between the pathways identified by GSVA and survival outcomes in patients with PDAC from the training cohort (Fig. 10D; P<0.05). Fig. 10D presents the results of the univariate Cox regression analysis. The horizontal axis represents the hazard ratio (HR) of the pathways, and the vertical axis lists the pathways identified by GSVA. The blue squares indicate the HR values, and the lines represent the 95% confidence intervals. Pathways with P<0.05 are considered significantly associated with survival outcomes.
Immune-related analysis
To evaluate the relationship between risk scores and the immune microenvironment in PDAC, immune cell infiltration levels were assessed using seven different algorithms: the ‘CIBERSORTx’ web portal (Legacy CIBERSORT August 2024 build; http://cibersort.stanford.edu/), the ‘MCPcounter’ R package (version 1.2.0; GitHub, http://github.com/ebecht/MCPcounter), the ‘EPIC’ R package (version 1.1.5; GitHub, http://github.com/GfellerLab/EPIC/releases/tag/v1.1), the ‘estimate’ R package (version 1.0.13; R-Forge, http://r-forge.r-project.org/projects/estimate/), the ‘TIMER’ web server (April 2025 build; http://timer.comp-genomics.org/), the ‘quanTIseq’ R script (commit 2024-10-15; GitHub, http://github.com/icbi-lab/quanTIseq), and the ‘xCell’ R package (version 1.1.0; CRAN, http://cran.r-project.org/package=xCell). The results were visualized using a heatmap, which was generated using the ComplexHeatmap package (version 2.13.1; Bioconductor, http://bioconductor.org/packages/ComplexHeatmap) (Fig. 11) Fig. 11 presents a comprehensive visualization of immune cell infiltration levels across different risk groups in PDAC, as assessed by seven distinct algorithms. The heatmap displays the relative abundance of various immune cell types, with rows representing specific immune cell populations and columns representing individual samples or risk groups. Each cell in the heatmap is color-coded to indicate the level of infiltration, ranging from low (blue) to high (red). The figure also includes annotations for risk scores and tumor types, allowing for the identification of patterns and correlations between immune cell infiltration and risk stratification. The clustering of samples based on immune infiltration profiles highlights potential differences in the immune microenvironment between high-risk and low-risk groups, providing insights into the immunological mechanisms underlying PDAC progression and prognosis.

Immune checkpoint and immune cell infiltration analysis
The expression of 46 immune checkpoints was analyzed in both groups, with statistical significance set at P<0.05 (Fig. 12A). Infiltration scores for 22 and 28 immune cell types were calculated using CIBERSORT and single sample GSEA, respectively, to estimate a diverse array of immune cell populations (Fig. 12B and C). Wilcoxon rank-sum tests were then applied to assess differences in immune cell infiltration between the groups (P<0.05). Furthermore, to assess the relationship between immune cell infiltration and prognostic genes, Spearman's correlation analysis was performed utilizing the Hmisc package. Immune cells exhibiting a correlation coefficient (|cor|) of >0.3 and P<0.05 were deemed to have a significant correlation. The relationships were illustrated in a correlation heatmap produced with the pheatmap package (Fig. 12D). Fig. 12D presents a correlation heatmap depicting the relationships between immune cells and Least Absolute Shrinkage and Selection Operator (LASSO)-selected genes. Each row represents a specific immune cell type, while each column corresponds to a LASSO-selected gene. The heatmap uses a color gradient to indicate the strength and direction of the correlations, with blue representing negative correlations and red representing positive correlations. Several immune cell types exhibit significant correlations with the prognostic genes, as indicated by the color intensity and the corresponding correlation coefficients. For example, certain immune cells show strong positive correlations (red blocks) with specific genes, suggesting potential synergistic roles in the immune response, while others display notable negative correlations (blue blocks), indicating possible antagonistic relationships. These findings highlight the complex interplay between immune cell infiltration and gene expression patterns in the context of PDAC, providing insights into the immune mechanisms that may influence disease progression and patient outcomes.
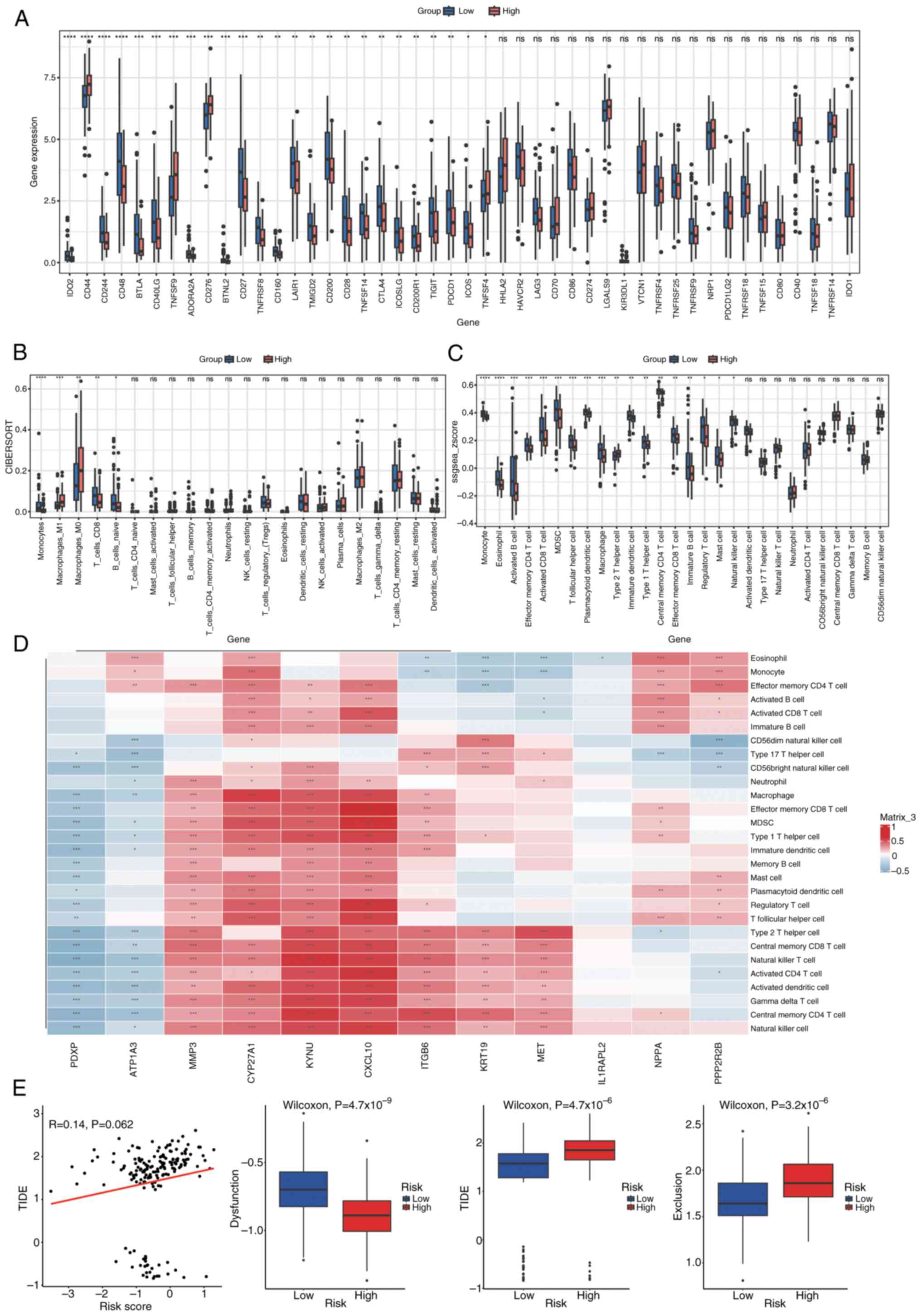
Assessment of treatment response in the high- vs. low-risk groups
The evaluation of treatment response differences involved the calculation of T cell dysfunction, T cell exclusion and the overall Tumor Immune Dysfunction and Exclusion (TIDE) score, employing the TIDE algorithm. The comparison of score distributions between the high- and low-risk groups was performed using Wilcoxon rank-sum tests (P<0.05; Fig. 12E). Fig. 12E illustrates the differences in dysfunction, exclusion and TIDE scores between high-risk and low-risk groups. A scatter plot shows the relationship between risk scores and TIDE scores, with each dot representing an individual sample. The red line indicates the trend of this relationship, suggesting a weak positive correlation between risk scores and TIDE scores (R=0.14, P=0.062), although the correlation is not statistically significant at the conventional threshold of P<0.05. Below the scatter plot, three box plots compare the distribution of dysfunction, TIDE and exclusion scores between the two risk groups. Each box plot displays the median, interquartile range (IQR) and potential outliers of the respective scores. The high-risk group shows significantly higher dysfunction scores (Wilcoxon test, P=4.7×10−9), TIDE scores (Wilcoxon test, P=4.4×10−6), and exclusion scores (Wilcoxon test, P=3.2×10−6) compared to the low-risk group. These results imply that the high-risk group may have more pronounced immune dysfunction and exclusion, which could contribute to adverse outcomes in patients with PDAC.
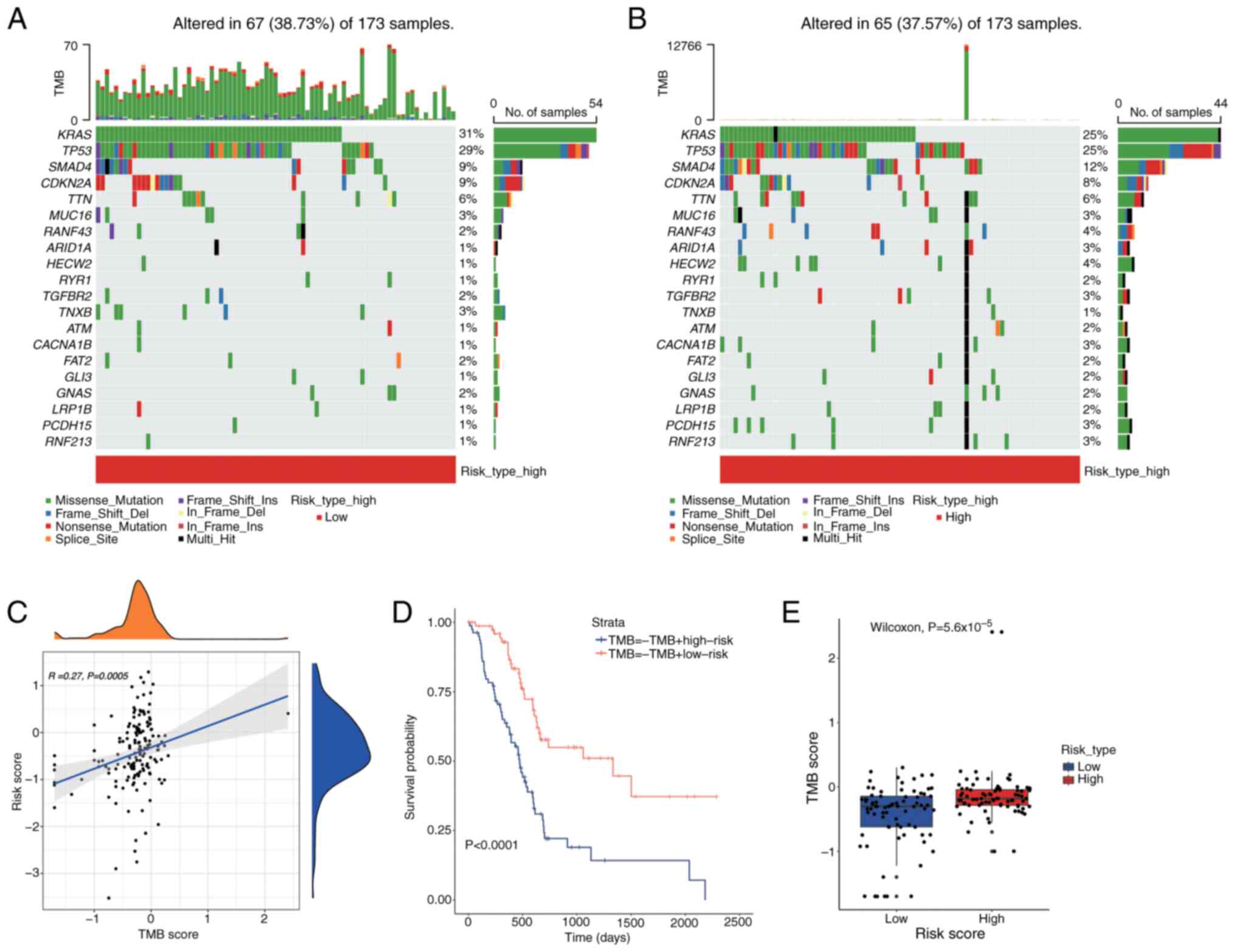
Tumor Mutational Burden (TMB) analysis
To assess the impact of TMB on PDAC prognosis, a waterfall plot was used to display the top 20 most frequently mutated genes across different risk categories (Fig. 13A and B). In the low-risk group, the APC gene demonstrated the highest mutation frequency at 25%, which increased to 31% in the high-risk group. The relationship between TMB and risk scores was illustrated using the tinyarray package (Fig. 13C). Fig. 13C illustrates the relationship between TMB scores and risk scores using a scatter plot and density plots. Each dot in the scatter plot represents an individual sample, with the x-axis indicating the TMB score and the y-axis indicating the risk score. A blue trend line shows a weak positive correlation between TMB scores and risk scores (R=0.27, P=0.0005), indicating that higher TMB scores tend to be associated with higher risk scores. The density plots on the top and right side of the scatter plot display the distribution of TMB scores and risk scores, respectively, showing the density of samples with different TMB and risk scores. Survival outcomes were assessed using Kaplan-Meier survival analysis utilizing the survival package to determine any differences across TMB levels (Fig. 13D). Fig. 13D presents a Kaplan-Meier survival analysis comparing the survival outcomes of different groups based on TMB and risk scores. The x-axis represents time in days, and the y-axis represents the survival probability. Two survival curves are shown: One for the TMB-high risk group (blue line) and one for the TMB-low risk group (red line). The results indicate a significant difference in survival between the groups, with the TMB-high risk group showing lower survival probabilities over time (P<0.0001). This suggests that patients with higher TMB and risk scores may have poorer prognoses. A Wilcoxon rank-sum test (P<0.05) was employed to analyze the differences in TMB scores across the groups (Fig. 13E). Fig. 13E displays a box plot comparing TMB scores between high-risk and low-risk groups. Each box plot shows the distribution of TMB scores within each group, with the box representing the IQR and the line inside the box indicating the median. The high-risk group exhibits significantly higher TMB scores compared to the low-risk group (Wilcoxon test, P=5.6×10−5), as evidenced by the higher median and overall distribution of TMB scores in the high-risk group.
Drug sensitivity analysis
To tailor individualized treatment strategies for PDAC, IC50 values for 198 drugs were calculated from the Genomics of Drug Sensitivity in Cancer database using the OncoPredict package. This analysis was performed on PDAC samples with comprehensive survival data from the training cohort. IC50 values, indicating drug sensitivity, were compared between the two groups using Wilcoxon rank-sum tests (P<0.05), and the top 10 most significant drugs were displayed in box plots (Fig. 14A). Additionally, Spearman correlation analysis, performed using the psych package, assessed the correlation between these drugs and risk scores (Fig. 14B), indicating a positive correlation for all drugs.
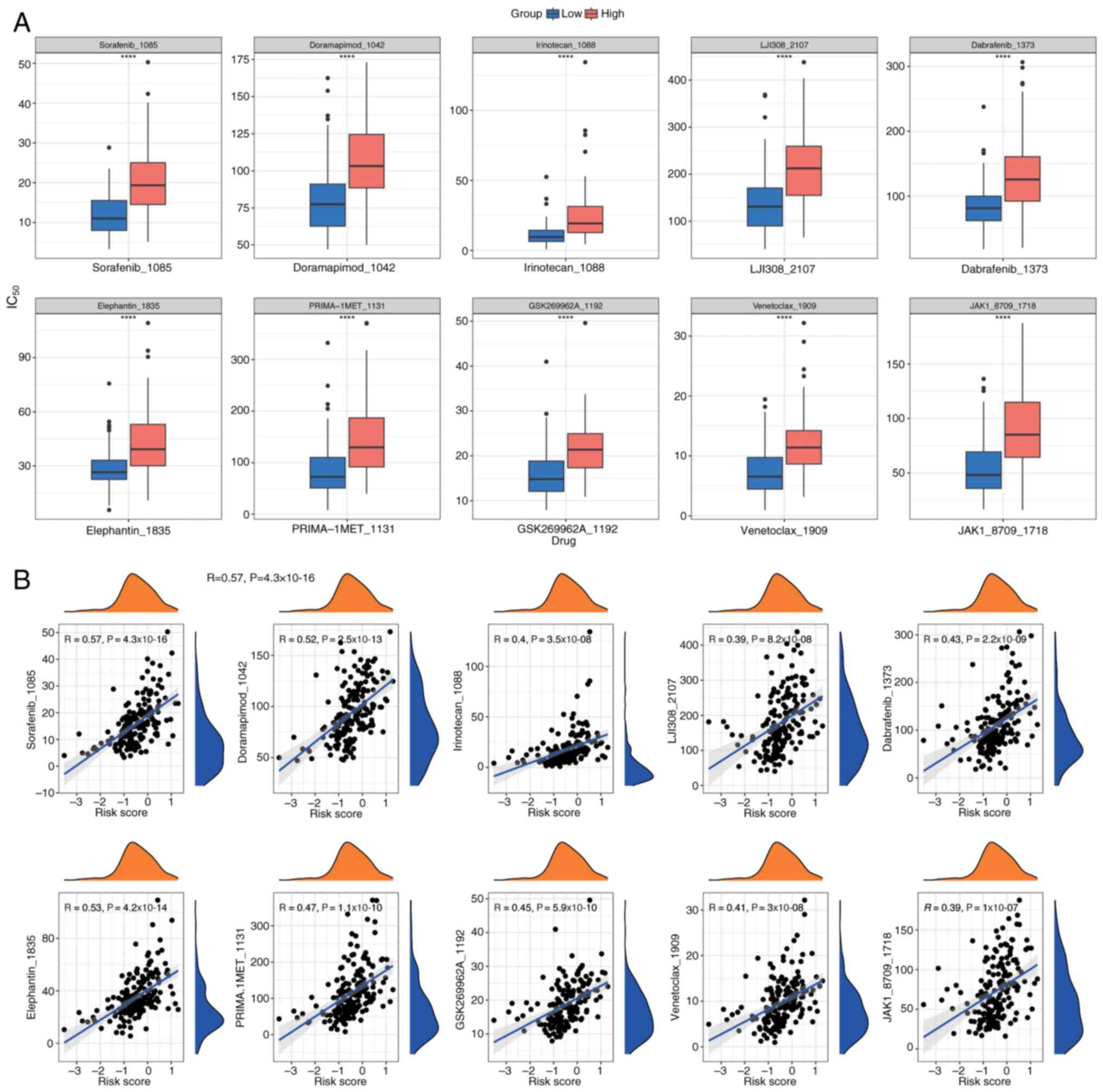
Prognostic gene correlation, localization, gene-gene interaction (GGI) network and regulatory network analysis
Spearman's correlation analysis was performed to evaluate the relationships among prognostic genes using the corrr package (Fig. 15A). Chromosomal localization of the biomarkers was analyzed using the RCircos package, which mapped their distribution across chromosomes: Natriuretic Peptide A (NPPA) on chromosome 1; KYNU, Integrin Subunit β6 (ITGB6) and CYP27A1 on chromosome 2; C-X-C Motif Chemokine Ligand 10 (CXCL10) on chromosome 4; Protein Phosphatase 2 Regulatory Subunit Bβ (PPP2R2B) on chromosome 5; MET on chromosome 7; Matrix Metalloproteinase 3 (MMP3) on chromosome 11; Keratin 19 on chromosome 17; ATPase Na+/K+ Transporting Subunit α3 on chromosome 19; Pyridoxal Phosphatase on chromosome 22; and Interleukin 1 Receptor Accessory Protein Like 2 on the X chromosome (Fig. 15B).
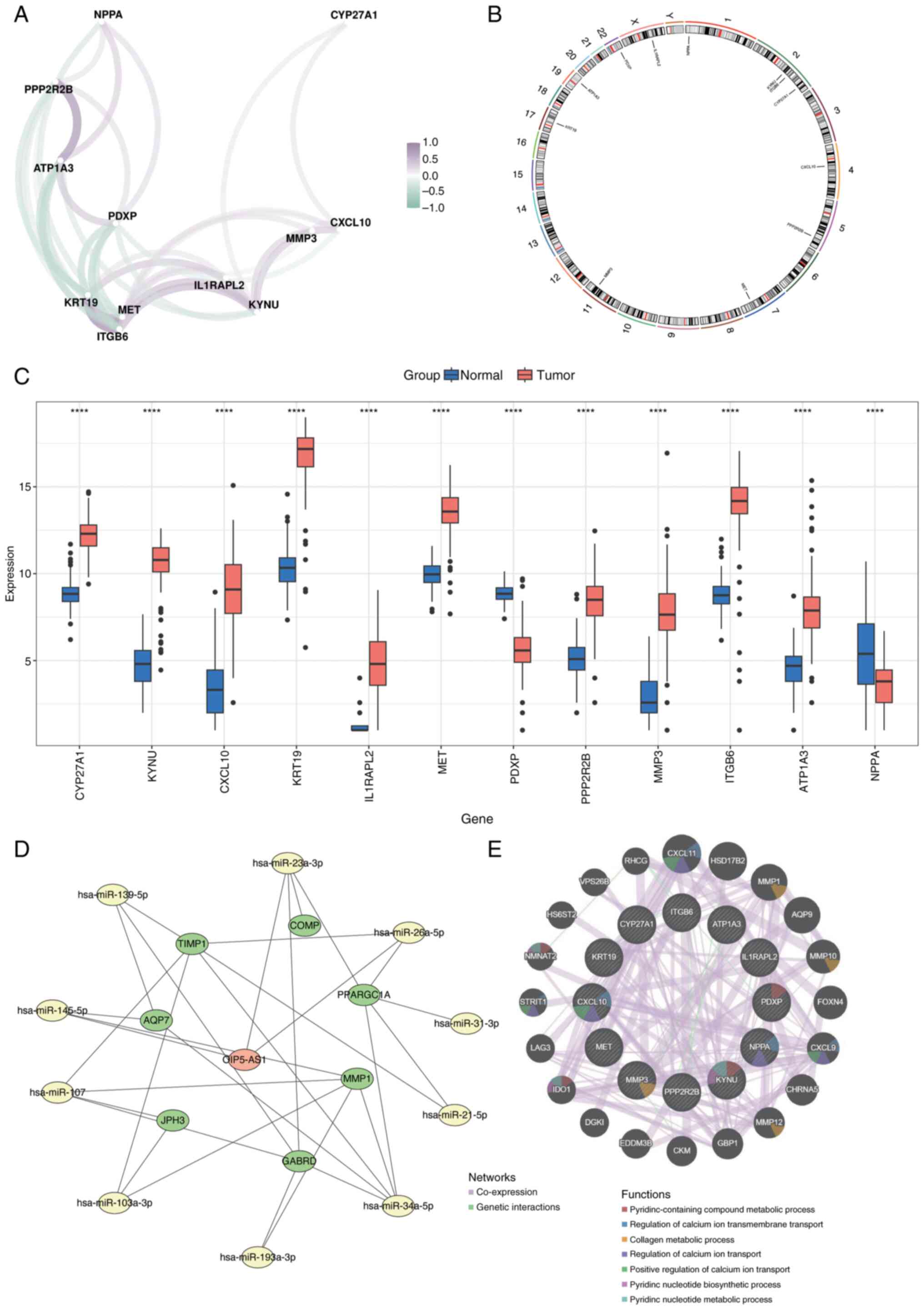
Expression levels of the 12 prognostic genes were compared between normal and cancerous tissues, revealing significant differences (P<0.05; Fig. 15C). To assess the molecular regulatory mechanisms underlying the prognostic genes, micro (mi)RNAs associated with these genes were predicted using the miRWalk and miRNet databases. miRNAs identified by both databases were selected as key candidates. Potential long noncoding (lnc)RNAs interacting with these miRNAs were then predicted using StarBase and miRNet. The intersecting lncRNAs identified by both databases were chosen as key lncRNAs. An lncRNA-miRNA-mRNA regulatory network was constructed with these key elements using Cytoscape (Fig. 15D). Additionally, the GeneMANIA database was utilized to identify genes functionally related to the prognostic genes and their associated biological functions. A co-expression network for these genes was created (Fig. 15E).
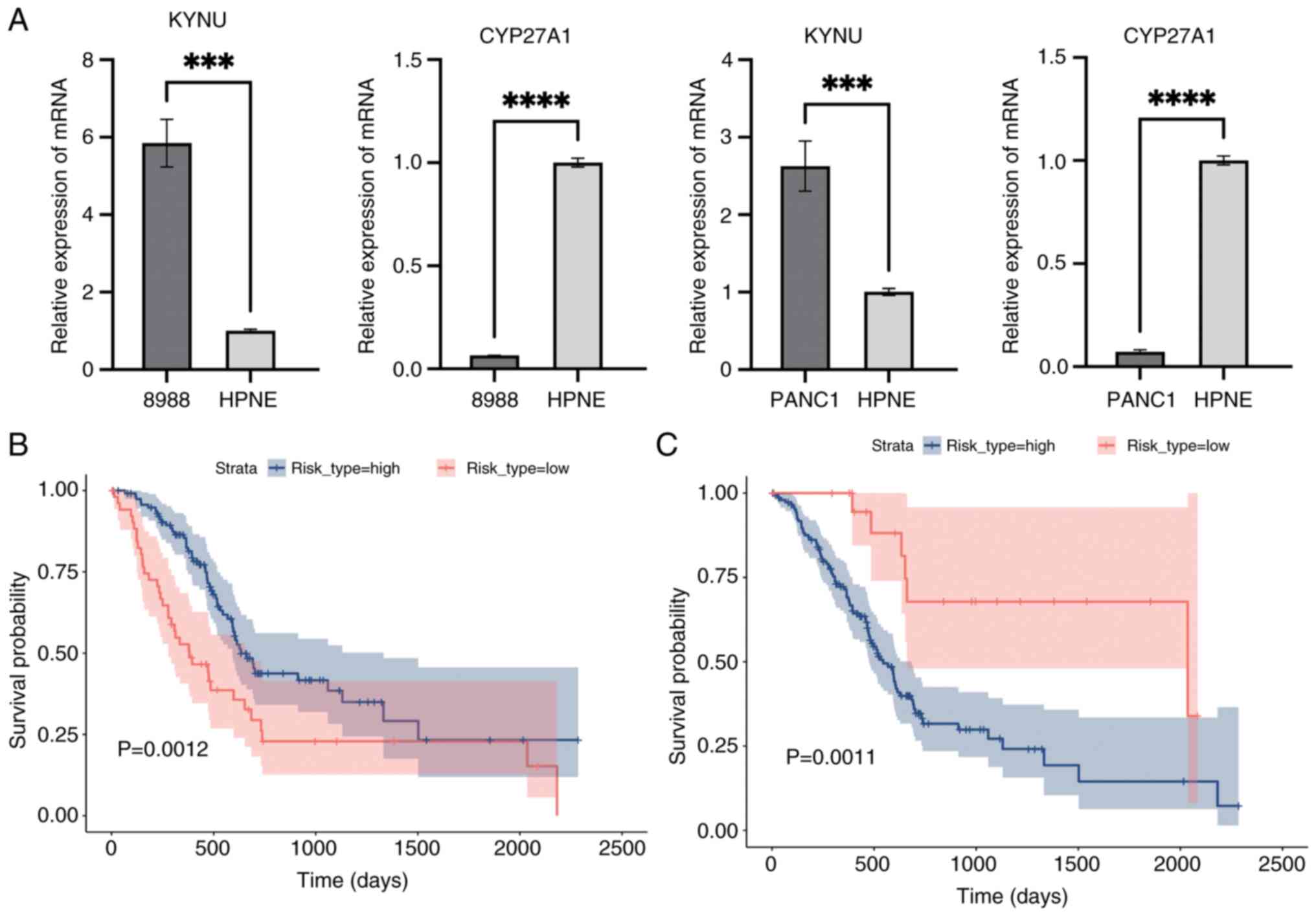
RT-qPCR and Kaplan-Meier validation
Median expression levels of CYP27A1, identified as a protective gene, and KYNU, recognized as a risk gene, were used to categorize the training cohort into high- and low-risk groups (Fig. 16A). Kaplan-Meier survival analysis then was performed to evaluate survival differences between these groups using the survival package, followed by log-rank tests to compare their survival outcomes (P<0.05; Fig. 16B and C). Fig. 16B and C present Kaplan-Meier survival curves comparing the survival outcomes of high-risk and low-risk groups based on the expression levels of CYP27A1 and KYNU. In both figures, the x-axis represents time in days, and the y-axis represents the survival probability. The blue line indicates the survival curve for the high-risk group, while the red line indicates the survival curve for the low-risk group. The shaded areas around the curves represent the 95% confidence intervals.
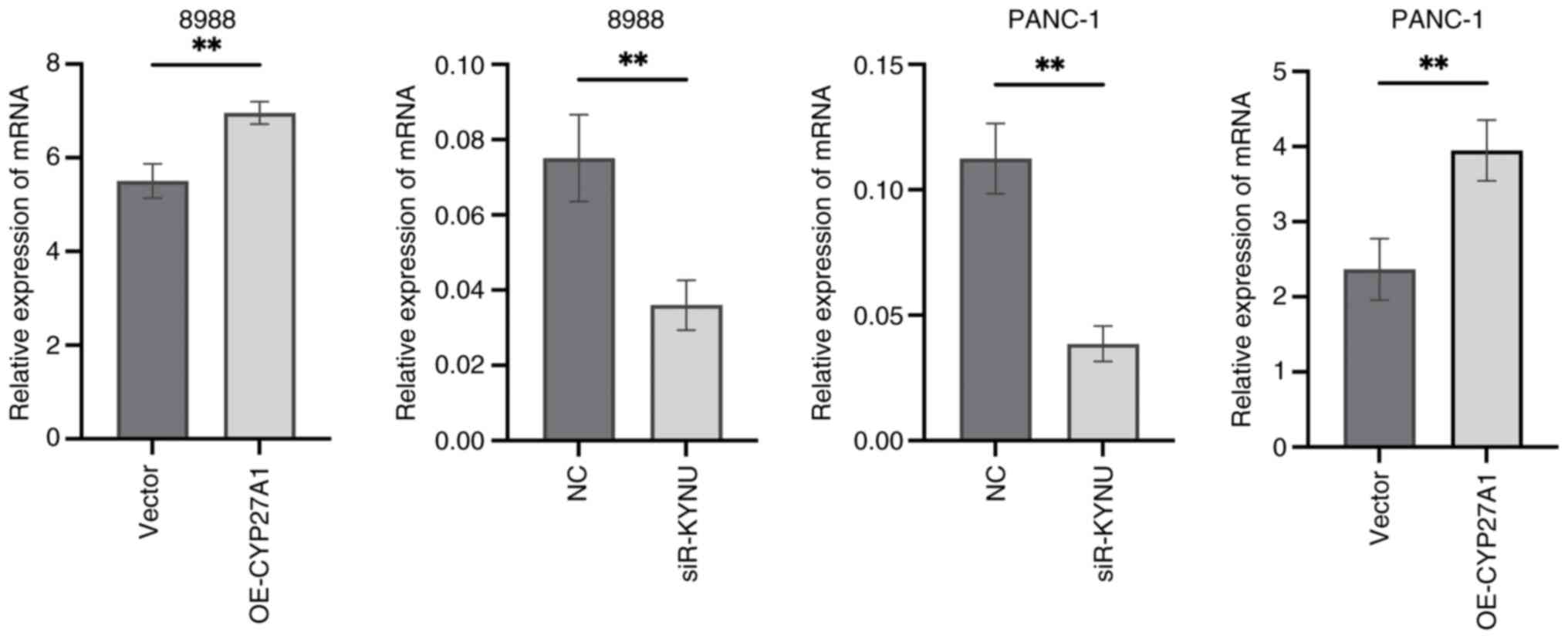
Validation of transfection efficiency
To assess the effectiveness of transfection, RT-qPCR results demonstrated that KYNU expression was significantly reduced in si-KYNU-transfected cells, whilst CYP27A1 expression was markedly increased following plasmid overexpression, compared with their respective controls (Fig. 17).
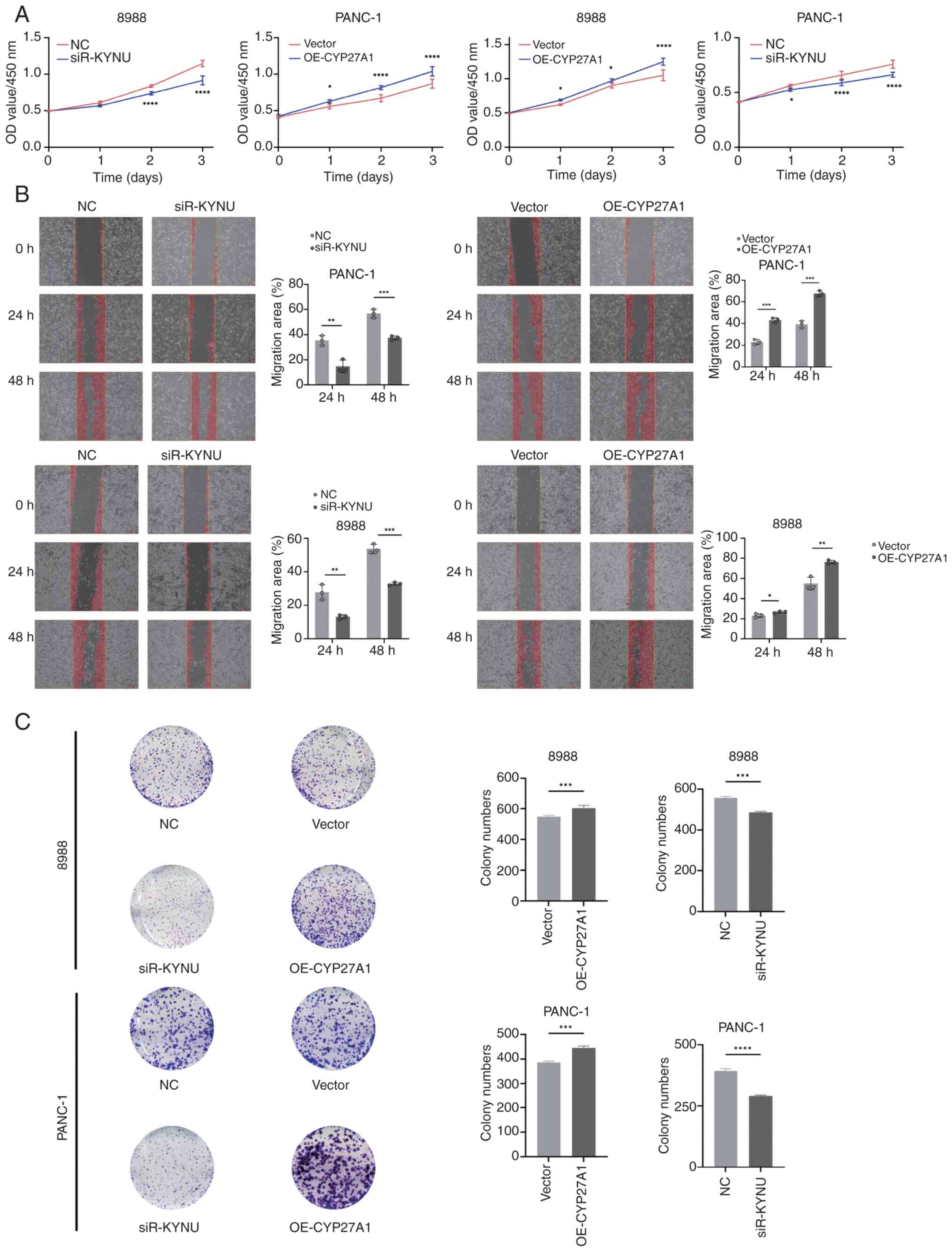
KYNU knockdown inhibits and CYP27A1 overexpression promotes PDAC proliferation and migration
CCK-8 assays were performed to assess the effects of KYNU knockdown and CYP27A1 overexpression on PDAC cell proliferation in 8988 and PANC-1 cells, revealing significant inhibition of proliferation with KYNU knockdown and enhanced proliferation with CYP27A1 overexpression compared with controls (Fig. 18A). WHAs indicated that KYNU knockdown in both cell lines significantly reduced migration rates at 24 and 48 h compared with the NC group, whilst CYP27A1 overexpression significantly increased the rate of migration in PANC-1 cells (Fig. 18B). Additional experiments in the two cell lines most affected by KYNU knockdown or CYP27A1 overexpression demonstrated that siR-KYNU significantly suppressed clonogenic capacity, whereas OE-CYP27A1 significantly increased proliferation compared with controls (Fig. 18C).
Discussion
The results of the GSEA analysis in the present study highlight the potential roles of Mn metabolism-related genes in PDAC pathogenesis. The enrichment of metabolic pathways such as ‘2-Oxocarboxylic acid metabolism’ in both the overall and downregulated gene sets suggests a disruption in core metabolic functions. Meanwhile, the enrichment of immune-related pathways in the upregulated gene set indicates that altered Mn metabolism may also influence immune responses and inflammation in PDAC. The interplay of these dual aspects highlights the intricate nature of Mn metabolism within tumor biology, laying the groundwork for subsequent investigations aimed at developing targeted therapeutic approaches. The present study employed bioinformatics techniques alongside transcriptomic data sourced from the GEO and TCGA databases to identify biomarkers associated with Mn metabolism that may serve as prognostic indicators for PDAC. Through the application of differential expression analysis, LASSO regression and multifactorial Cox regression models, 12 significant genes were identified that exhibit a strong correlation with the prognosis of PDAC. The validation of these prognostic genes, performed with both the training cohort and external datasets (GSE62452 and GSE28735), reinforced the robustness and clinical relevance of the model in the present study, which demonstrated AUCs of 0.82 and 0.83 for the training and external validation sets, respectively. The results provide valuable understanding of the molecular mechanisms at play in PDAC and underscore the possible role of Mn metabolism in the advancement of this aggressive cancer type.
Mn, a vital trace element, is recognized for its critical involvement in numerous biological functions, such as regulating OS, mediating cellular signaling and supporting metabolic pathways. Serving as a cofactor for enzymes like MnSOD, Mn is crucial for maintaining cellular integrity by counteracting ROS and minimizing OS (31). Imbalances in Mn metabolism, whether caused by deficiency or excess, can disrupt vital biological pathways, resulting in cellular stress and potentially contributing to the onset of cancer (32,33).
The present study contributes to the growing body of evidence suggesting that Mn metabolism is disrupted in several cancers, including PDAC. For example, studies have indicated that Mn deficiency can activate the tumor suppressor protein p53, leading to mitochondrial-induced apoptosis and genomic instability (34–36). Similarly, excess Mn has been associated with neurodegenerative diseases, but its role in cancer progression is becoming a topic of increasing interest. Previous work has highlighted Mn as a critical player in modulating immune responses and TME composition, which are critical factors in PDAC progression (37).
The genes identified in the present study are involved in several BPs crucial to PDAC pathogenesis, including cell adhesion, extracellular matrix remodeling, immune modulation and inflammation. For example, CXCL10, a chemokine that mediates immune cell recruitment, has been reported to regulate the PDAC microenvironment. It can facilitate tumor progression by attracting immune cells like T-cells and natural killer cells, which serve a dual role in either controlling or exacerbating tumor growth depending on the context (38). The analysis in the present study identified CXCL10 as a crucial biomarker, underscoring its potential role as both a prognostic marker and a possible therapeutic target for PDAC.
MMP3 is another significant gene recognized in the present analysis. MMP3 serves a crucial role in the degradation of the extracellular matrix (ECM) and is associated with notable processes in cancer development, such as tumor invasion, metastasis and epithelial-to-mesenchymal transition, which is vital for the metastatic dissemination of PDAC (39,40). The upregulation of MMP3 expression has been associated with adverse prognoses in multiple cancer types, notably PDAC (40). Its involvement in the remodeling of the ECM further validates its role as a significant feature gene in the predictive model in the present study.
MET, a receptor tyrosine kinase, serves a critical role in regulating cellular processes such as proliferation, survival and migration by interacting with its ligand, hepatocyte growth factor. Alterations in MET signaling pathways have been frequently reported in numerous cancer types, including PDAC, where it contributes to tumor progression, invasiveness and metastatic spread (41,42). In the present study, MET was identified as a crucial biomarker, reflecting its established involvement in PDAC. This finding highlights the potential of targeting MET with inhibitors, which are currently being evaluated in clinical trials (42).
The immune microenvironment is fundamental in the development of PDAC, a type of cancer well-known for its capacity to escape immune detection. The immune-related analyses in the present study emphasize the importance of the immune system in PDAC prognosis, particularly through the influence of the identified biomarkers on immune cell infiltration and tumor immune escape. Recent studies indicate that the PDAC immune microenvironment is characterized by T-cell dysfunction and expansion of myeloid-derived suppressor cells, both of which substantially contribute to therapeutic resistance (43). The genes identified in the present study may serve as important mediators in these processes and provide valuable insights into how Mn metabolism influences immune modulation in PDAC.
Moreover, the gene CYP27A1, which encodes a cytochrome P450 enzyme involved in sterol metabolism, was demonstrated to be differentially expressed in PDAC tissues. Although its precise function in cancer remains unclear, earlier research emphasized its possible role in regulating immune responses via cholesterol metabolism processes (44). This indicates that disruptions in lipid metabolism, common in cancer cells, may also affect immune cell function and contribute to PDAC progression (45).
In addition to gene expression analysis, a GGI network was constructed to predict functional relationships between the identified biomarkers and their potential regulatory mechanisms. This analysis identified several key interactions that may clarify how these genes collectively contribute to PDAC pathogenesis. For example, NPPA has been associated with the regulation of blood pressure and vascular function, and recent studies have reported its involvement in the regulation of tumor angiogenesis (46–50). PPP2R2B, a regulatory component of protein phosphatase 2A, is involved in several signaling cascades, particularly those pertaining to cell cycle regulation and programmed cell death. This suggests its potential contribution to sustaining the survival of tumor cells in PDAC (51). These findings underscore the complexity of the regulatory networks that govern PDAC progression and highlight the need for further investigation into how these pathways intersect.
Although the present research provides new perspectives on the prognostic significance of genes related to Mn metabolism in PDAC, certain limitations must be acknowledged. Whilst the risk model in the present study demonstrated a robust performance in independent validation, its applicability to non-Western populations requires further study due to potential genetic and environmental heterogeneity. One of the key limitations is that the findings are primarily based on transcriptomic data, meaning the predictive model developed may not entirely capture the intricate biological characteristics of PDAC. The expression of RNA does not consistently align with the levels of protein or its functional activity. Additionally, post-translational modifications, including processes like phosphorylation and glycosylation, can alter the functional roles of the genes identified (52,53). Therefore, experimental validation using techniques such as western blotting, immunohistochemistry and mass spectrometry is essential to confirm the expression and functional relevance of these biomarkers in PDAC tissues. Furthermore, although external validation datasets (GSE62452 and GSE28735) were used to evaluate the accuracy of the predictive model, the diversity present in public datasets may introduce variability in the outcomes. This variability may stem from differences in sample collection methods, patient populations and technical platforms used, which can introduce biases that affect the generalizability of the findings. Future studies should aim to validate the identified biomarkers in large, independent cohorts and in clinical settings. Finally, the molecular mechanisms underlying Mn metabolism in PDAC remain incompletely understood. Although the present study provides valuable information about gene expression patterns and potential pathways, the interaction between Mn metabolism, OS and immune modulation requires further exploration. By incorporating multi-omics strategies, including proteomics, metabolomics and single-cell RNA sequencing, a deeper and more holistic insight can be gained into the role of Mn metabolism in shaping PDAC biology across several layers (54,55). Prospective collaborations with multi-institutional cohorts across diverse geographical regions will be essential to validate and refine the biomarker panel in the present study.
In summary, the results of the present research offers insights into the involvement of Mn metabolism-associated genes in the prognosis of PDAC. The 12 biomarkers identified through bioinformatics analysis provide a promising foundation for developing a prognostic risk model for PDAC, which could assist in patient stratification and guide treatment decisions. Furthermore, the biological functions and potential regulatory mechanisms of these biomarkers illuminate the complex processes involved in PDAC progression, highlighting the need for further experimental validation. By exploring the interplay between Mn metabolism, immune modulation and cancer progression, the present research opens new avenues for targeted therapies and precision medicine in PDAC.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and Technology Commission of Shanghai Municipality (grant nos. 19401935000 and 20Y21901600) and the Shanghai ‘14th Five-Year Plan’ Chinese Medicine Specialty Construction Project (grant no. ZYTSZK2-2)
Availability of data and materials
The datasets analyzed during the current study are available in the following repositories. TCGA-PDAC dataset: TCGA. Validation datasets: GEO under accession numbers GSE62452 and GSE28735. Drug sensitivity data (IC50 values) were obtained from the GDSC database (https://www.cancerRxgene.org/), which requires registration for access. The custom data generated in the present study may be requested from the corresponding author.
Authors' contributions
SC and SL contributed to the conceptualization and design of the study and revised the manuscript. ZX performed data analysis and drafted the manuscript. ZZ performed data analysis, interpreted the results and assisted in manuscript writing. All authors read and approved the final manuscript. ZX and ZZ confirm the authenticity of all the raw data.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Siegel RL, Miller KD, Fuchs HE and Jemal A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74:2913–2921. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Chintamani Singhal V: Temporary closure of open abdominal wounds by the modified sandwich-vacuum pack technique (Br J Surg 2003; 90: 718–722). Br J Surg. 90:1452–1453. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 381:317–327. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Raghavan D: Introduction: Bladder cancer. Semin Oncol. 39:5232012. View Article : Google Scholar : PubMed/NCBI | |
|
Li D, Xie K, Wolff R and Abbruzzese JL: Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al: Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Kanji ZS, Edwards AM, Mandelson MT, Sahar N, Lin BS, Badiozamani K, Song G, Alseidi A, Biehl TR, Kozarek RA, et al: Gemcitabine and taxane adjuvant therapy with chemoradiation in resected pancreatic cancer: A novel strategy for improved survival? Ann Surg Oncol. 25:1052–1060. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al: Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Grant TJ, Hua K and Singh A: Molecular pathogenesis of pancreatic cancer. Prog Mol Biol Transl Sci. 144:241–275. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Duan H, Li L and He S: Advances and prospects in the treatment of pancreatic cancer. Int J Nanomedicine. 18:3973–3988. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Liu J, Ji S, Liang C, Qin Y, Jin K, Liang D, Xu W, Shi S, Zhang B, Liu L, et al: Critical role of oncogenic KRAS in pancreatic cancer (review). Mol Med Rep. 13:4943–4949. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Sun H, Zhang B and Li H: The roles of frequently mutated genes of pancreatic cancer in regulation of tumor microenvironment. Technol Cancer Res Treat. 19:15330338209209692020. View Article : Google Scholar : PubMed/NCBI | |
|
Hafezi S, Saber-Ayad M and Abdel-Rahman WM: Highlights on the role of KRAS mutations in reshaping the microenvironment of pancreatic adenocarcinoma. Int J Mol Sci. 22:102192021. View Article : Google Scholar : PubMed/NCBI | |
|
George B, Kudryashova O, Kravets A, Thalji S, Malarkannan S, Kurzrock R, Chernyavskaya E, Gusakova M, Kravchenko D, Tychinin D, et al: Transcriptomic-based microenvironment classification reveals precision medicine strategies for pancreatic ductal adenocarcinoma. Gastroenterology. 166:859–871.e3. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Bear AS, Vonderheide RH and O'Hara MH: Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell. 38:788–802. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al: PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Baj J, Flieger W, Barbachowska A, Kowalska B, Flieger M, Forma A, Teresiński G, Portincasa P, Buszewicz G, Radzikowska-Büchner E and Flieger J: Consequences of disturbing manganese homeostasis. Int J Mol Sci. 24:149592023. View Article : Google Scholar : PubMed/NCBI | |
|
Lin M, Colon-Perez LM, Sambo DO, Miller DR, Lebowitz JJ, Jimenez-Rondan F, Cousins RJ, Horenstein N, Aydemir TB, Febo M and Khoshbouei H: Mechanism of manganese dysregulation of dopamine neuronal activity. J Neurosci. 40:5871–5891. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Yanjun Y, Jing Z, Yifei S, Gangzhao G, Chenxin Y, Qiang W, Qiang Y and Shuwen H: Trace elements in pancreatic cancer. Cancer Med. 13:e74542024. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao N, Zhang AS, Wortham AM, Jue S, Knutson MD and Enns CA: The tumor suppressor, P53, decreases the metal transporter, ZIP14. Nutrients. 9:13352017. View Article : Google Scholar : PubMed/NCBI | |
|
Konzack A, Jakupovic M, Kubaichuk K, Görlach A, Dombrowski F, Miinalainen I, Sormunen R and Kietzmann T: Mitochondrial dysfunction due to lack of manganese superoxide dismutase promotes hepatocarcinogenesis. Antioxid Redox Signal. 23:1059–1075. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN and Jiang X: Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 572:402–406. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Saleh SAK, Adly HM, Abdelkhaliq AA and Nassir AM: Serum levels of selenium, zinc, copper, manganese, and iron in prostate cancer patients. Curr Urol. 14:44–49. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Wang W, Chen G, Zhang W, Zhang X, Huang M, Li C, Wang L, Lu Z and Xia J: The crucial prognostic signaling pathways of pancreatic ductal adenocarcinoma were identified by single-cell and bulk RNA sequencing data. Hum Genet. 143:1109–1129. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Xu X, Peng Q, Jiang X, Tan S, Yang Y, Yang W, Han Y, Chen Y, Oyang L, Lin J, et al: Metabolic reprogramming and epigenetic modifications in cancer: From the impacts and mechanisms to the treatment potential. Exp Mol Med. 55:1357–1370. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou Z, Cheng Y, Jiang Y, Liu S, Zhang M, Liu J and Zhao Q: Ten hub genes associated with progression and prognosis of pancreatic carcinoma identified by co-expression analysis. Int J Biol Sci. 14:124–36. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Xiang XH, Mao J and Li H: Expression and clinical significance of centromere protein A in pancreatic cancer based on GEO and TCGA database analysis. Chin J Pancreatol. 20:184–189. 2020.(In Chinese). | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Sahoo K and Sharma A: Understanding the mechanistic roles of environmental heavy metal stressors in regulating ferroptosis: Adding new paradigms to the links with diseases. Apoptosis. 28:277–292. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Rozenberg JM, Kamynina M, Sorokin M, Zolotovskaia M, Koroleva E, Kremenchutckaya K, Gudkov A, Buzdin A and Borisov N: The role of the metabolism of zinc and manganese ions in human cancerogenesis. Biomedicines. 10:10722022. View Article : Google Scholar : PubMed/NCBI | |
|
Diessl J, Berndtsson J, Broeskamp F, Habernig L, Kohler V, Vazquez-Calvo C, Nandy A, Peselj C, Drobysheva S, Pelosi L, et al: Manganese-driven CoQ deficiency. Nat Commun. 13:60612022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang J and Chen X: p53 tumor suppressor and iron homeostasis. FEBS J. 286:620–629. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos D, et al: Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. 57:860–872. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Alaimo A, Gorojod RM, Miglietta EA, Villarreal A, Ramos AJ and Kotler ML: Manganese induces mitochondrial dynamics impairment and apoptotic cell death: A study in human Gli36 cells. Neurosci Lett. 554:76–81. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Stelling MP, Soares MA, Cardoso SC, Motta JM, de Abreu JC, Antunes MJM, de Freitas VG, Moraes JA, Castelo-Branco MTL, Pérez CA and Pavão MSG: Manganese systemic distribution is modulated in vivo during tumor progression and affects tumor cell migration and invasion in vitro. Sci Rep. 11:158332021. View Article : Google Scholar : PubMed/NCBI | |
|
Pandey V, Fleming-Martinez A, Bastea L, Doeppler HR, Eisenhauer J, Le T, Edenfield B and Storz P: CXCL10/CXCR3 signaling contributes to an inflammatory microenvironment and its blockade enhances progression of murine pancreatic precancerous lesions. Elife. 10:e606462021. View Article : Google Scholar : PubMed/NCBI | |
|
Gobin E, Bagwell K, Wagner J, Mysona D, Sandirasegarane S, Smith N, Bai S, Sharma A, Schleifer R and She JX: A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer. 19:5812019. View Article : Google Scholar : PubMed/NCBI | |
|
Luan H, Jian L, Huang Y, Guo Y and Zhou L: Identification of novel therapeutic target and prognostic biomarker in matrix metalloproteinase gene family in pancreatic cancer. Sci Rep. 13:172112023. View Article : Google Scholar : PubMed/NCBI | |
|
Fu J, Su X, Li Z, Deng L, Liu X, Feng X and Peng J: HGF/c-MET pathway in cancer: From molecular characterization to clinical evidence. Oncogene. 40:4625–4651. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Guo R, Luo J, Chang J, Rekhtman N, Arcila M and Drilon A: MET-dependent solid tumours-molecular diagnosis and targeted therapy. Nat Rev Clin Oncol. 17:569–587. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Nie Y, Liu C, Liu Q and Zhu X: CXCL10 is a prognostic marker for pancreatic adenocarcinoma and tumor microenvironment remodeling. BMC Cancer. 23:1502023. View Article : Google Scholar : PubMed/NCBI | |
|
He S, Ma L, Baek AE, Vardanyan A, Vembar V, Chen JJ, Nelson AT, Burdette JE and Nelson ER: Host CYP27A1 expression is essential for ovarian cancer progression. Endocr Relat Cancer. 26:659–675. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Niu N, Shen X, Wang Z, Chen Y, Weng Y, Yu F, Tang Y, Lu P, Liu M, Wang L, et al: Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell. 42:869–884.e9. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, Guiducci C, Kathiresan S, Benjamin EJ, Struck J, et al: Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet. 41:348–353. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Forte M, Marchitti S, Di Nonno F, Stanzione R, Schirone L, Cotugno M, Bianchi F, Schiavon S, Raffa S, Ranieri D, et al: NPPA/atrial natriuretic peptide is an extracellular modulator of autophagy in the heart. Autophagy. 19:1087–1099. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Song P, Yao X, Zhong T, Zhang S, Guo Y, Ren W, Huang D, Duan XC, Yin YF, Zhang SS and Zhang X: The anti-tumor efficacy of 3-(2-nitrophenyl) propionic acid-paclitaxel (NPPA-PTX): A novel paclitaxel bioreductive prodrug. Oncotarget. 7:48467–48480. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Song W, Wang H and Wu Q: Atrial natriuretic peptide in cardiovascular biology and disease (NPPA). Gene. 569:1–6. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Luan Y, Xie B and Wei W: REST-repressed lncRNA NPPA-AS1 regulates cervical cancer progression by modulating miR-302e/DKK1/wnt/β-catenin signaling pathway. J Cell Biochem. 122:16–28. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Malvi P, Chava S, Cai G, Hu K, Zhu LJ, Edwards YJK, Green MR, Gupta R and Wajapeyee N: HOXC6 drives a therapeutically targetable pancreatic cancer growth and metastasis pathway by regulating MSK1 and PPP2R2B. Cell Rep Med. 4:1012852023. View Article : Google Scholar : PubMed/NCBI | |
|
Li J, Zhang Y, Yang C and Rong R: Discrepant mRNA and protein expression in immune cells. Curr Genomics. 21:560–563. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Ramazi S and Zahiri J: Posttranslational modifications in proteins: Resources, tools and prediction methods. Database (Oxford). 2021:baab0122021. View Article : Google Scholar : PubMed/NCBI | |
|
Nalbantoglu S and Karadag A: Metabolomics bridging proteomics along metabolites/oncometabolites and protein modifications: Paving the way toward integrative multiomics. J Pharm Biomed Anal. 199:1140312021. View Article : Google Scholar : PubMed/NCBI | |
|
Gautam SK, Batra SK and Jain M: Molecular and metabolic regulation of immunosuppression in metastatic pancreatic ductal adenocarcinoma. Mol Cancer. 22:1182023. View Article : Google Scholar : PubMed/NCBI |










