



HPV and cervical cancer: From molecular diagnostics to emerging treatment approaches (Review)
- Authors:
- Published online on: April 22, 2025 https://doi.org/10.3892/wasj.2025.346
- Article Number: 58
-
Copyright : © Kabir et al. This is an open access article distributed under the terms of Creative Commons Attribution License [CC BY 4.0].
Abstract
1. Introduction
Cervical cancer is the fourth most common type of cancer globally, one of the leading causes of mortality and the second most common type of cancer among women; the average age at diagnosis is 53 years, and the average age at death is 59 years (1,2). An estimated 604,127 cases and 341,831 related deaths were recorded globally in 2020, with South Central Asia having the highest incidence of 148,128 cases (3). China and India contributed to the majority of the global cervical cancer burden with 97,000 cases and 60,000 related deaths in India, and 106,000 cases and 48,000 related deaths in China (1). The incidence of cervical cancer is disproportionately higher in less developed countries, possibly due to high infection rates with infectious agents, poor access to screening, public health priorities, cultural barriers, technology, healthcare infrastructure and the high cost of human papillomavirus (HPV) vaccines in such countries (4). In developed western countries however, the incidence of cervical cancer is decreasing due to improvements in screening and treatment modalities, in addition to lifestyle modifications. In the USA, the incidence of cervical cancer has significantly decreased since the 1970s due to implementation of the Papanicolaou screening test. Despite this, an estimated 13,800 cases and 4,290 deaths from cervical cancer occurred in 2020 in the USA (5,6). While in 2025, 13,360 new cases and 4320 death are estimated to occur (7). The current global burden of cervical cancer based on the Global Cancer Observatory (GLOBOCAN) 2022, reported that an estimated 662,044 cases and 348,709 death occurred in 2022(8). Cervical cancer continues to be the leading cause of cancer-related mortality that results in 10 premature deaths weekly among women aged 20-39 years (9). Geographical location is a major factor that has been implicated for variations in cervical cancer screening, with women residing in urban areas having a higher likelihood of being screened than those in rural areas (10). Other barriers to screening may be both personal or associated with structural factors in accessing care. Personal barriers may include the lack of awareness on risk factors and screening modalities, cultural influence, embarrassment and anxiety, as well as the fear of being diagnosed with cancer. On the other hand, structural barriers may include cost, language barriers and the lack of physicians or access to physicians (10). Cervical cancer is caused by HPV, particularly types 16 and 18, and to a lesser degree, 10 other types of the virus, as reported by the International Agency for Research on Cancer (IARC) (2,11). The majority of HPV types cause asymptomatic infection of the skin (cutaneous type) and anogenital tract (mucosal), which may later progress to cervical, anal or head and neck cancer (12).
Cervical cancer is not a new disease; it has been studied as part of women's history and the history of gynecology. For the past two millennia, cervical cancer has been in existence as the archetypal cancer among women due to its specificity, frequency and unpleasant symptoms, such as uncontrolled vaginal bleeding, offensive discharge and severe pain. However, not until the mid-19th century, a clear pathological definition of cancer was not developed and recognized in modern medicine. In the 1830s, the application of a microscope to the study of normal and abnormal tissues and cells ushered a precise definition of cervical cancer, even though there were numerous concerns raised about its subjectivity, reproducibility, as well as clinical utility. However, with further advances, the definition of cancer is now firmly based on microscopic pathology, not on symptoms or clinical examination (13).
Over the past 40 years, HPVs are a key causative agent of cervical cancer; however, the phylogenetic analysis into different genomes of HPVs has proven that the virus family can be traced back millions of years. Of note, >200 genotypes of HPV have been identified to date and are grouped into low-risk HPV (LR-HPV) and high-risk HPV (HR-HPV) based on their oncogenic capability over the past four decades. To date, ~15 HR-HPV have been identified with a worldwide prevalence of 10% and a higher prevalence of 22% in Africa, with the prevalence decreasing with an increase in age. However, women with persistent infection are at higher risk of developing high-grade squamous intraepithelial lesion (HSIL) squamous cell carcinoma (SCC) (1,10-12). In addition, HPV has been implicated in males with a prevalence ranging from 1 to 7%, and a lower rate of HPV clearance has been observed in uncircumcised males than in those how have been circumcised (8). It has been shown that HR-HPVs are associated with cervical intraepithelial neoplasia (CIN), invasive cervical cancer, and its precursor lesions in women, whereas in males, they are associated with penile cancer, and head and neck SCC (1,8).
Considering the global burden of cervical cancer and as this is a significant cause of morbidity and mortality amongst women, despite being a vaccine-preventable disease, the elucidation of the mechanisms underlying its development will help improve its diagnosis and treatment. Thus, the present review aimed to provide an overview of the association between HPV infection and the development of cervical cancer, while highlighting the molecular mechanisms involved in cervical cancer progression, such as polymorphism and immune evasion by HPV. The diagnostic and treatment approaches to cervical cancer will also be discussed.
2. Anatomy of the cervix
The cervix, as well as the upper part of the vagina, the body of the uterus and fallopian tubes are all derived from the paramesonephric duct. The cervix is the most inferior part of the uterus, with a length of ~2.5 cm. Superior to the cervix is the body of the uterus, and inferior to it is the vagina. The cervical canal communicates with the vagina and uterus through the external os and internal os, respectively. The lower part of the cervix projects into the anterior vaginal wall and divides into two parts, i.e., the vaginal part and supravaginal part. The spaces between the former and the vagina form the vaginal fornices. The latter is related to the bladder anteriorly, rectovaginal pouch and rectum posteriorly, and on either side to the ureter and uterine artery embedded in the parametrium. The cervix is supplied by the uterine artery, drained by the external iliac, internal iliac, obturator and sacral lymph nodes, and innervated by sympathetic (T12-L2), parasympathetic (S2-S4) and viscerosensory nerves. Histologically, the cervix is mainly composed of fibroelastic connective tissue and is divided into ectocervix, squamocolumnar junction and endocervix. The ectocervix is made of squamous epithelium, while the endocervix is made of columnar epithelium. The junction between these two parts represents the squamocolumnar junction and serves as the point cervical cancer originates (9).
3. Cervical cancer biology
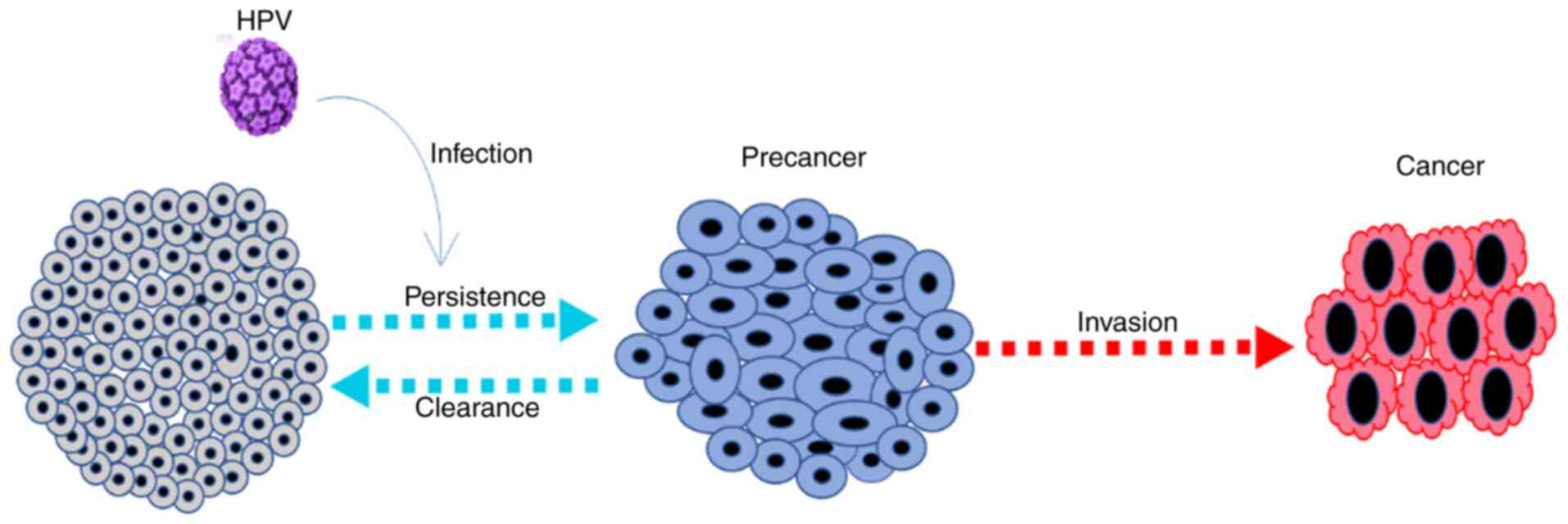
Cervical cancer arises from the normal cervical epithelium, which undergoes progressive changes from low to high-grade CINs over years. HPV infects epithelial cells via interaction with cell surface receptors such as integrin α6 and integrates its DNA into the basal cells of the squamocolumnar junction, thus resulting in the production of proteins that causes CIN. CIN may progress to cancer or regress (Fig. 1). In the case of progression, a virus with a low copy number increases to ~50-100 copies/cell following DNA replication. The DNA replication is initiated by E1 and E2 proteins when they bind to the viral origin resulting in the assembly of E1-E2 ternary complex, thus preventing nonspecific interaction of E1-DNA, serving as a template for the recruitment of additional molecular binding of E1 and E2 and assembly of the E1 double-trimer intermediate. This is followed by the unwinding of the DNA, expression of viral oncogenes (E6 and E7) and capsid proteins (L1 and L2), followed by the assembly of multiple copies of infectious viruses that infect neighboring cells and eventually cause dysplasia (8,9). It is important to note that not all women who become infected with HPV develop CIN or cervical cancer, as the immune system clears off most of the infection.
The squamocolumnar junction zone is composed of squamous and columnar glandular epithelium that arises from adaptive conversion known as metaplasia. This adaptation results from trauma and the acidic pH of the vagina, resulting in increased production of chemical mediators that interfere with antiviral immune response, hence enhancing HPV infection (16).
Metaplastic cells have an altered maturation and weak cell envelope and intermediate filaments, hence having less resistance to mechanical stress leading to cell death. These immature cells are more vulnerable to HPV infection due to both the basal cells and basement membrane are more accessible in metaplastic areas due to their monostratified nature (17).
Increased levels of estrogen and progesterone receptor-positive cells are found in squamous metaplastic cells than normal squamous cells. Sex hormones through steroid response elements stimulate the expression of E6 and E7 genes, hence facilitating malignant progression (18). Some hormones also alter immune responses leading to persistent HPV infection. The hormonal status of women not only enhances the maintenance and development of metaplastic cells, but also leads to high sensitivity of the transformation zone to HPV infection and malignant progression.
Metaplastic cells of the transformation zone not only promote HPV infection, but also aid the development of anal and cervical carcinoma when the immune response is undermined. However, HPV lesions at anogenital sites other than the transformation zone (vulva and vagina) are not likely to progress to malignancy. Hence physiological, immunological and anatomical nature of the transformation zone not only enhances viral entry but also enhances HPV-induced malignant progression (17).
Polymorphisms and cervical cancer
Genetic polymorphism is linked with the development of cervical cancer and other cancer It has been suggested that variants of the same HPV types are biologically different, and hence may result in distinct pathogenic abilities (19). Mutations in the E2 binding site result in the negative regulation of the P97 promoter, relieving E2-mediated promoter repression, leading to increased transcription of E6 and E7 genes, hence increasing HPV immortalization ability. In addition, polymorphisms in human leukocyte antigen (HLA), p53, pRB, CYP1A1, MBL2 gene exon1, TGFBT10C, TGFBc509T, FasR-1377G, CCR2V6L, HLADRB1 and IL-10-1082G/A genes have all been shown to be associated with an increased risk of developing cervical cancer (20).
The interaction of the HPV E6 protein with the Arg allele of p53 at codon 72 leads to its degradation in the ubiquitin-proteasome pathway. Breast cancer susceptibility gene 1 (BRCA1) is antagonized by the E6 protein (21). Another study reported that individuals with BRCA1 mutation have an increased risk of developing cervical cancer, with BRCA1 Pro871leu polymorphism highly implicated in cervical carcinogenesis (22).
4. Biology of HPV
HPV is an 8-kb non-enveloped double-stranded DNA (dsDNA) virus with a circular genome having a diameter of 55 nm. The dsDNA molecule is contained in a protein capsid comprised of major (L1) and minor (L2) capsid proteins. The capsid contains 72 capsomeres, each comprised of five monomers of 55 kDa that join to form the L1 pentameric protein. These L1 pentamers and copies of L2 proteins join to form an icosahedral symmetry of the virion. Each HPV genome is composed of 8 open reading frames (ORFs) transcribed from a single DNA strand. The ORFs are made of three functional regions, i.e., the early (E) region, the late (L) region and the long control region (LCR). LCR is a non-coding region responsible for the transcription and replication of viral DNA. The E region consists of E1, E2, E4, E5, E6 and E7 proteins that are involved in viral replication and oncogenesis, while the L region encodes the structural proteins L1 and L2 that are involved in viral assembly (12,14).
Pathogenesis of HPV in cervical cancer
The pathogenesis of HPV infection can be discussed in the context of viral persistence and clearance. A viral clearance occurs after 12-18 months of infection by a humoral and cell-mediated immune response directed towards the same HPV type. Viral clearance may indicate an absence of HPV antigens, antibodies or DNA using a sensitive assay method (16).
Infection with HPV is considered to be persistent if the virus is detected within 4-6 months in two consecutive follow-ups. This persistence may increase the risk of developing malignant lesions resulting from centrosomal and chromosomal abnormalities, leading to the accumulation of genetic abnormalities over a period of time, thus allowing the development of cancer (23). However, differentiating persistent infection from healing followed by re-infection is confusing; some studies have shown that an infection is termed persistent if the same HPV genotype is detected in two consecutive follow-ups within 4-6 months. Even with this information however, some infections may be assumed to be cleared due to low detectable levels, which will later resurface before the next follow-up (24).
Genomic instability, such as centrosomal abnormalities induced by E6 and E7 contributes to the persistent nature of HPV infection and the subsequent progression to malignancy (23,25). There is an increased demand for nutrients and oxygen in HPV lesions. The formation of tetraploid cells by inducing cell fusion and failure of cytokinesis are essential functions of the E5 protein in HPV infection and tumor progression. E6 induces HPV-related malignancies through p53 ubiquitination by the formation of ubiquitin ligase complex (UBE3A). The expression of E7 triggers increased levels and the expression of hypoxia-inducible factor-1, resulting in increased angiogenesis, thus favoring the persistence and growth of HPV lesions (14,23).
The association between HPV and the development of cervical cancer follows a pattern: Sexual exposure, persistent infection, the development of pre-cancerous lesions and the progression to invasive disease (26). However, recently, non-sexual transmission has been reported to occur through the following mechanisms: i) Horizontal transmission by direct contact with the pathogen through contaminated clothes, utensils, environment or surgical equipment; ii) vertical transmission from mother to newborn; iii) self-inoculation of oral warts to the genitalia; and/or iv) water-bone transmission (27).
Immune system activation by HPV in cervical cancer
Upon reaching the epithelial basal layer microlesions, HPV is sometimes prevented from entering into keratinocytes due to the acidic pH of the cervical mucosa and the presence of mucoproteins. However, most often, HPV evades these protective mechanisms and infects the keratinocytes. Following this, the innate immune response is initiated to trigger a rapid inflammatory response. This response involves the recruitment of immune cells, such as natural killer (NK) cells, NK T-cells, dendritic cells and Langerhans cells (LCs) at the infection site (28).
Immune activation begins with keratinocytes enclosing replicating HPV as non-professional antigen-presenting cells (APCs) that present viral peptides in MHC-I. Even though keratinocytes do not express MHC II, which is essential in the control and accumulation of T helper 1 (Th1) cells, they can be stimulated with interferon γ (IFN-γ) or other cytokines to do so. Keratinocytes can also express different pattern recognition receptors, such as Toll-like receptors, and can produce cytokines, such as TNF-α, IL-18 and chemokines, such as CCL2 and CXCL9, all of which facilitate the recruitment and activation of more immune cells such as macrophages, dendritic cells, NK cells, CD4+ and CD8+ cells (29,30). On the other hand, mucous epithelial layers have professional APCs that are essential for activating the adaptive immune system, leading to B-cell activation and the production of antibodies. These professional APCs comprising of dendritic cells and macrophages captures and present the HPV-infected cells to T-cells, in addition to expressing pattern recognition receptors capable of activating and expressing cytokines necessary for activating CD4 and CD8 T-cells (31). The activation of the Toll-like receptors by non-methylated viral DNA triggers both innate and adaptive immune response that creates a pro-inflammatory environment and promotes cytokine production with the aim of finally eliminating the infection.
Immune evasion mechanism
Chronic HPV infection that persists for months, or even years may result in the host immune system becoming unaware or used to the virus (Fig. 2). This will render the host immune response insufficient in clearing the virus, and thus an infected individual develops persistent infection (32). This occurs as HPV DNA replication and assembly occur in terminally differentiated cells as means of evading the host immune response (33).
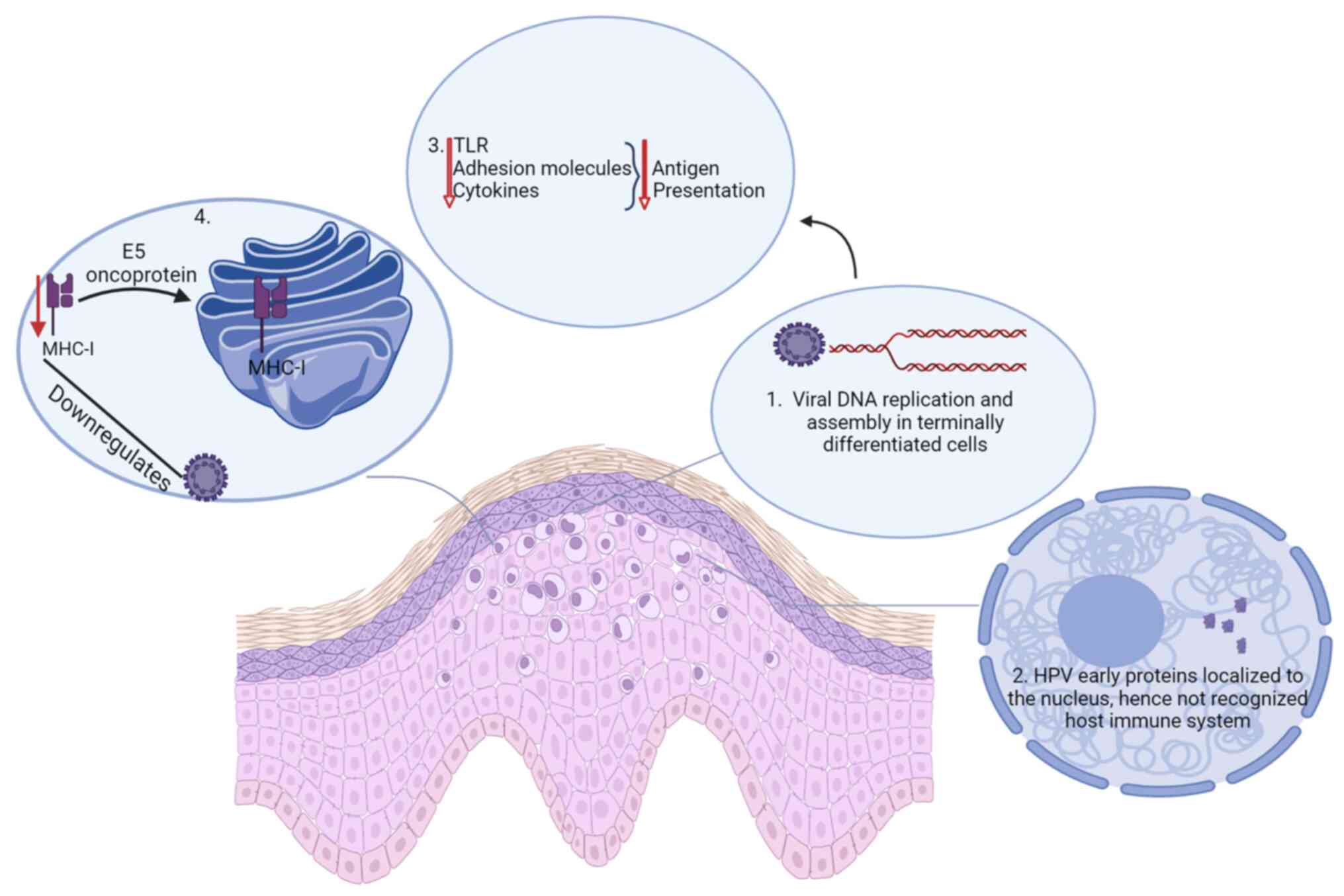
HPV early proteins are produced in insufficient amounts and are also localized to the nucleus, thus not accessible for immune recognition (14,34). Cytokines, adhesion molecules and Toll-like receptors are in most stages of the viral infectious cycle not released, leading to decreased or lack of antigen presentation, and thus absence of signals that will initiate immune response in the epithelial cells. In addition, HPV downregulates the expression of MHC-I on the cell surface by retaining it in the Golgi apparatus orchestrated by the E5 oncoprotein (14,33).
HPV replication and release do not result in inflammation and cell death as the terminally differentiating keratinocytes are programmed to die by natural causes, and thus, the absence of signals that will trigger an inflammatory response reduces the ability of the host to mount an effective host response, thus making the virus to appear invisible to the host immune system (32). HPV evades the innate immune response by minimizing its exposure to the immune system. It utilizes the keratinocyte differentiation process to complete its replication cycle within the superficial layers involved virion assembly, thus facilitating its release through the natural desquamation process mediated by the E4 protein, without triggering an immune response (35). Furthermore, the expression of capsid proteins in differentiated epithelium is minimized by HPV, leading to delayed or lack of detection by LCs, which in turn results in persistent infection.
Another immune evasion mechanism used by HPV is the absence of T-cell activation receptors and costimulatory molecules responsible for the identification and elimination of transformed cells. CD40 is an example of this, and its expression has been reported to be promoted by the transcriptional factor of the AT-Hook AKNA type. However, the effect of HPV infection results in decreased CD40 and IL-8 activation, thereby influencing the development and maintenance of adaptive immune response (36).
Despite NK cells being key effectors of the early immune surveillance of tumors, their activity is controlled by the balance between inhibition and activation receptors. In a particular study, the loss of MHC-I caused by viral oncoproteins was expected to result in the susceptibility of the tumor cells to the cytotoxic effects of NK cells (31). However, the patients exhibited dysfunctional NK cells that were unable to exert an effective cytotoxic effect on the tumor. This dysfunction was reported to be possibly due to defects in the expression of some important activation receptors, such as NKp30 and NKp46 that were observed to decreased in peripheral NK cells of the cervical cancer patients, suggesting that defects in activation receptors may promote cervical cancer progression (31,37).
5. Cervical cancer risk factors
Despite HPV DNA being present in almost all cervical cancers, a multitude of other risk factors are involved the development of the disease, unlike other types of cancer that have a small number of associated risk factors. The immune state of an individual is a major determining factor for a persistent HPV infection. Immunocompromised women with human immunodeficiency syndrome (HIV) or on chronic immunosuppressive therapy are at a higher risk of developing invasive cervical cancer. Smoking is also a major risk factor that increases the likelihood of developing cervical cancer by two-fold. Nicotine and other tobacco products have been reported to damage cervical epithelial cells and impair the local immune response (38,39). The risk of developing cervical cancer increases proportionately with the duration of oral contraceptives use (40).
Multiple pregnancies are also associated with an increased risk of developing cervical cancer. Women who have had over five pregnancies have a two-fold higher risk of developing cervical cancer than those with only one or two pregnancies (40). Hormonal imbalance during pregnancy may increase the susceptibility of HPV infection, in addition to a weakened immune system. In addition, women with multiple sex partners have been reported have a three-fold increased risk of developing cervical cancer than those with only one sex partner. In addition, women who had their first parity at an age <17 years also have a three-fold increased risk of developing cervical cancer (41).
Women with limited access to Pap smear testing due to personal or systemic factors are also at higher risk of developing cervical cancer as they are less likely to be diagnosed and treated for pre-invasive cervical lesions (10).
6. Cervical cancer diagnosis
The diagnosis of cervical cancer is primarily associated with assessing the cervical epithelium through regular screening by Pap smear, colposcopy and viral DNA detection. By applying these methods, the onset and development of cervical cancer can be arrested as early as possible. Cervical cancer develops gradually through a series of processes from mild neoplasia (CIN1), through more severe degrees of neoplasia (CIN2 or CIN3), to invasive disease. These stages of development have been the basis for diagnosis and treatment. It has been observed that different HPV types are associated with varying degrees of neoplasia, with each neoplasia being a distinct process. CIN1 is usually observed as a self-limiting sexually transmitted HPV infection that is more likely to regress, while CIN2 and CIN3 are regarded as the true precursors of cervical cancer. The risk of progression from mild to severe dysplasia is only 1% per year, while the risk of progression from mild to severe dysplasia is 16% within 2 years, and 25% within 5 years (42,43). Despite this, it is considered that early detection and the treatment of HPV can prevent the progression of precancerous lesions to cancer.
Cervical cancer diagnosis is not a ‘one size fits all’ approach due to heterogeneity of the disease with respect to risk factors, immune status, lifestyle or socioeconomic status, race, and most importantly, the age of the patient. For asymptomatic infections, such as cervical neoplasia, and/or routine screening, Pap screening is recommended for women aged 21 years. HPV DNA testing and Pap smears are recommended for women aged 22-30 years, with a Pap screening every 3 years for those with negative results and at a low risk of developing cervical cancer. For those between 30-65 years of age, a Pap smear should be performed after every 5 years with HPV DNA testing. For women >65 years of age, and with continued negative screening, both cytology and HPV DNA testing should be discontinued (44).
In cases of abnormal cytological results and/or persistent HPV-infection, a colposcopy is the procedure of choice, as recommended by the American Society for Colposcopy and Cervical Pathology (ASCCP). The algorithms recommended by the society are considered the standard of care. This includes multiple colposcopic-guided biopsies and endocervical sampling (except in pregnancy) (45). For patients with invasive disease however, the International Federation of Gynecology and Obstetrics (FIGO) staging system is recommended. This system is based on the local extent of the tumor that is determined with a combination of laboratory tests, pelvic examination, proctoscopy, cystoscopy, chest X-ray, intravenous pyrography, and PET and MRI scans (44).
Pap smear
The primary screening method for the detection of early cervical epithelial changes and the early stage of invasive cervical cancer is the Pap smear test. This method was first introduced by George N. Papalicolaou in 1949, even before HPV was identified as the causative of cervical cancer (42). This method looks for changes in cells of the squamocolumnar junction. The reporting guidelines for Pap smear have evolved, from the CIN system (which is based solely on tissue architecture) to the Bethesda system. The Bethesda system however, was developed to reflect an advanced understanding of cervical neoplasia while introducing uniform descriptive diagnostic terminologies, in addition to including a statement on specimen adequacy as an integral part of the report (43). This system classifies squamous cell neoplasia into four categories: Atypical squamous cell of undetermined significance (ASCUS), atypical squamous cells with a possibility of a high-grade squamous intraepithelial lesion (ASC-H), low-grade squamous intraepithelial lesions (LSIL), HSIL and SCC.
The ASCUS represents a borderline that includes lesions that have cellular abnormalities suggestive of a squamous intraepithelial lesion, ASC-H represents cells also do not appear normal but could be at higher risk of being preneoplastic compared with ASCUS, LSIL include mild neoplasia usually due to transient HPV infection, while HSIL represents moderate to severe cervical intraepithelial dysplasia and carcinoma in situ.
Despite some limitations associated with Pap smears, such as sample inadequacy, false-negative results, contamination associated with sample collection, interobserver error and sample processing error, there has been a significant reduction in mortality from cervical cancer due to widespread screening programs, especially in developed countries. The overall sensitivity of the Pap test in detecting an HSIL is 70.8%. To achieve maximum sensitivity for early detection of precancerous lesions, Pap screening is done in association with the HPV DNA test (46).
Visual inspection with acetic acid (VIA)
This method is carried out based on the ability of acetic acid to dehydrate abnormal cells that contains increased nuclear material and protein. Following the application of 5% acetic acid to the cervix, abnormal areas turn white, while healthy cells containing glycogen retain their normal color. Similarly, Lugol's iodine can be employed for the same purpose. However, in this case, healthy cells containing glycogen take up iodine and appear brown, while abnormal cells remain unstained. In both methods, the abnormal areas can be biopsied and examined. Dull white plaques with uneven borders are considered LSIL, while those with thick and sharp edges are considered HSIL (15). Despite having low specificity, this method has shown less false-negative rate and can be a good alternative, particularly in low-resource areas where Pap screening is not available.
Colposcopy
Colposcopy is the magnified visualization of the cervix usually in association with VIA. It is performed on patients with abnormal Pap smear findings, but without a gross cervical lesion. In the case that abnormal areas are identified, the areas are biopsied with the aid of a colposcope. A colposcope is a binocular instrument that has a magnifying power of x10-20. The cervix is exposed with a speculum, while the patient is lying in a lithotomy position. The colposcope is placed at a distance of 20 cm and focused on the external os. The cervix is then swabbed with normal saline to remove excess mucus without triggering bleeding and then visualized by VIA. A satisfactory colposcopic examination is one in which both columnar epithelium, squamous epithelium and squamocolumnar junction are visualized (15,42). However, in the case that the entire squamocolumnar junction cannot be visualized, a cone biopsy is carried out. The tissues collected can be examined histologically by observing characteristic pathologic features, such as hyperchromasia, increased nucleo-cytoplasmic ratio, acanthosis, koilocytosis in terminally differentiated keratinocytes with atypical nuclei, stromal inflammation, stromal desmoplastic reaction, loss of nuclear polarity, numerous single or clusters of dysplastic cells and elongated rete ridges.
HPV DNA testing
This method detects infection with HR-HPV by examining samples obtained similar to Pap smear either using a spatula or cytobrush and then stored in a viral transport medium, or collected into a liquid-based cytology container. The collected samples are then used for the detection of HR-HPV DNA or mRNA following nucleic acid extraction and polymerase chain reaction (PCR). This method is recommended for sexually active women >30 years of age and can be used as a first-line testing method or in combination with cytology.
HPV DNA testing can be in the form of type-specific PCR or a general primer PCR. The former targets a single HPV type by utilizing the variations present in the E6 and E7 genes of each HPV subtype. The latter however, has the capability of amplifying more than 15 HPV types in a single PCR run by targeting the conserved L1 gene using consensus primers such as MY09/MY11 and GP5+/GP6+ (2,15,42). After the PCR run, the amplified HPV genotypes can be identified either by restriction fragment length polymorphism (RFLP), sequencing, or by hybridization with type-specific probes.
7. Cervical cancer treatment
The management of cervical cancer and other premalignant lesions is carried out according to the findings. Premalignant lesions are managed cautiously for women <25 years of age and in the majority of cases, the majority of findings are low-risk cervical dysplasia, which usually resolves spontaneously. However, lesions suspected to be greater than low-risk, are evaluated by colposcopy, low-risk legions are observed and re-evaluated more frequently, while high-risk lesions are treated based on staging, size and location. For premalignant lesions that are limited in size and depth, excision or cryotherapy is done, while conization or the loop electrosurgical excision procedure is performed for lesions that have invaded the endocervical canal and beyond. For early-stage invasive cancer, the treatment is surgical resection either by conization or radical hysterectomy; however, women in this category who still require pregnancy are treated using a trachelectomy. Women with high-risk features post-resection may be subjected to adjuvant therapy with radiotherapy and/or chemotherapy.
Surgery
Surgical resection is performed for patients with early-stage disease that is restricted to the cervix. This can range from non-invasive techniques, such as cervical conization to radical hysterectomy. Cervical conization is indicated for patients with carcinoma in situ or stage IA1 invasive cervical cancer, and it is performed with a cold knife cone (CKC) to remove the squamocolumnar junction and ~3 mm margin of the cervix. Pathological evaluation of the presence of lymphovascular invasion (LVI) and margins of the lesion is essential, and if present, further surgical intervention may be required. But if no LVI is present, nodal evaluation is not necessary. Patients that have undergone this procedure mostly have a 5-year survival rate >95% (44).
Radical trachelectomy is indicated for patients who desire to preserve their fertility. This requires the removal of the majority of the cervix (leaving ~5 mm), and resection of the parametria. The 5 mm of the cervix is preserved to enable the placement of a cerclage for future pregnancy. Due to the increased risk of nodal involvement, sentinel lymph node biopsy or pelvic lymphadenectomy mostly accompanies radical trachelectomy. In case of adverse pathologic features adjuvant treatment with radiotherapy with or without chemotherapy is necessary.
Radical hysterectomy is indicated for patients who do not desire to preserve their fertility. Four types of hysterectomy have been defined, with type A representing only a minimal parametrial resection, type B representing resection of the paracervix at the level of the ureter, type C representing resection of the paracervix at the level of the internal iliac vessels, and type D representing resection of the paracervix to the pelvic sidewall. This procedure however, is associated with complications, such as infection, hemorrhage, small bowel obstruction, vesicovaginal fistula, venous thromboembolism and pulmonary embolus.
Another form of surgery indicated for patients with stage IA1 who do not also desire to preserve their fertility is an extrafascial hysterectomy. This involves the removal of the entire cervix and uterus without removal of the parametria, while removal of the ovaries is optional. This technique may be done either by opening the abdominal cavity (laparotomy) or through the vagina. Complete parametrectomy or radiotherapy may be done post-operatively if adverse pathologic features are discovered.
Radiotherapy
Radiotherapy is a treatment modality for cervical cancer as a definitive or adjuvant therapy with or without chemotherapy. For early-stage cervical cancer IA1-IIA1, radiotherapy may be used as the sole treatment modality. However, definitive concurrent platinum-based chemoradiotherapy with brachytherapy boost is essential for advanced cervical cancer. Post-operative radiotherapy with or without chemotherapy is essential when surgical pathologic conditions are observed. In post-radical hysterectomy patients, at least two of the following criteria must be met when using adjuvant radiotherapy without chemotherapy i.e., the tumor size must be >4 cm, there must be greater than one-third stromal invasion, or lymphovascular space invasion (LVSI) (44).
Radiotherapy is delivered either as brachytherapy or external beam radiotherapy (EBRT). The EBRT is composed of intensity-modulated radiotherapy (IMRT) and is directed at the primary and pelvic lymphatics, while Brachytherapy involves delivering radiation close to the tumor using a sealed radiation source either alone in early-stage lesions or as a boost following EBRT in more advanced lesions. A high dose rate of >12 Gy/h generated from a radioactive source (usually Iridium-192) is applied for a maximum of 8 weeks.
Immunotherapy
Multiple molecular features, such as microsatellite instability (MSI), high tumor inflammatory state, high tumor mutational burden (TMB) and a high expression of programmed death ligand 1 (PD-L1) supports the use of immunotherapy in cervical cancer treatment (47). MSI, an established predictor for response to immunotherapy in various cancer reported to be present in 8% of cervical cancers (48). TMB was also established as an independent predictor of treatment outcome with immune checkpoint inhibitors (ICIs). PD-L1 expression downregulates T-cell activity, thereby providing an immune privileged site for the establishment and persistence of HPV infection, as reported in 35-95% of cervical cancer (49).
Immunotherapy is currently considered an effective treatment strategy that guarantees successful recovery. Promising results have been obtained with this approach, with various classes of compounds being studied, such as tumor infiltrating lymphocytes, antibody-drug conjugate and vaccines (prophylactic and therapeutic). Among these, the most widely studied immunotherapy compounds are the ICI. Most importantly, ICIs with PD-1 and cytotoxic T-lymphocyte antigen 4 (CTLA4) as the most commonly targeted molecules are being explored in cervical cancer treatment (47). CTLA4, a homolog of CD28 that have a higher affinity for B7, regulates effector T cells. T-cells are activated majorly by the binding of CD28 to B7 on APCs, and this is achieved only when there is CD28-B7 level less than that of CTLA4-B7. On the other hand, PD-1, a costimulatory receptor commonly found in exhausted T-cells due to prolonged stimulation caused by chronic inflammation or cancer, prevents the production of TCR signaling and decreases the production of IL-2, IFNγ, and TNF-α when it binds to PD-L1(50).
A successful antitumor response is dependent upon an effective processing and the presentation of tumor antigen by dendritic cells or activated APCs, resulting in the recognition of the antigen by unique T-cell receptors, thereby providing the initial signal for T-cell activation to be implemented by CD28-B7. This, in addition to a subset of CD8+ T-cells that differentiate into effector T-cells, kills all cells displaying the tumor antigen. However, to ensure long-term immunity, the effector T-cells must differentiate into memory T-cells, depending on the interaction with dendritic cells and CD4+ helper T-cells (51).
Resistance to ICIs may arise as a result of high tumor mutational burden due to the increased production of neoantigens, or lack of their recognition. In addition, genetic and epigenetic alterations may lead to the disruption or loss of the MHC-1 and/or B2M molecules that are responsible for antigen processing and presentation mechanisms even in tumors with high antigenicity (47). It should be noted that a combination approach of using ICIs and other strategies, such as radiotherapy, chemotherapy and other strategies that improve antigen presentation, or block immunosuppressive pathways are essential for preventing resistance to immunotherapy.
Another evolving immunotherapy approach involves the use of NK cells to exert a direct cytotoxic effect on tumor cells by releasing perforins and granzymes. They can also indirectly kill tumor cells by producing cytokines such as IFN-γ, which in turn activate other cells of the immune system. However, this is a promising area that needs to be elucidated separately.
HPV vaccines
Vaccination is targeted towards preventing HPV infection; however, the public health goal is preventing cervical cancer. HPV vaccination is recommended for both males and females <15 years of age; even though males cannot develop cervical cancer, the vaccine will reduce the risk of the transmission of the virus from these vaccinated males to their female sexual partners (15,52). Vaccination appears to be a cost-effective measure of preventing microbial and viral-induced diseases. However, the prophylactic HPV vaccines, Cervarix, Quadrivalent Gardasil and Gardasil-9, which are readily available, are not yet included in the national immunization programs of a number of countries; furthermore, these vaccines are relatively costly, thus making ‘out-of-pocket’ purchases almost impossible for populations in low and middle-income countries.
The aforementioned vaccines are termed prophylactic HPV vaccines. These include viral-like particles synthesized from the L1 capsid protein of HPV, and have the same antigenicity as the virion, thereby eliciting an immune response against future infection with HPV. Cervarix is established from the L1 capsid protein of HPV 16 and 18, quadrivalent Gardasil from the L1 capsid protein of HPV 6, 11, 16, and 18, while Gardasil-9 is established from the L1 capsid protein of HPV 6, 11,16, 18, 31, 33, 45, 52 and 58.
Despite being able to provide protection against HPV infection, prophylactic HPV vaccines however have some limitations. The first limitation is that vaccination with any of these vaccine types will render protection only against the HPV types from which that vaccine is comprised of. Therefore, infection with other HPV types will not be neutralized by the host immunity. Another limitation is that these vaccines are not effective in eliminating a pre-existing HPV-induced lesion as the virus has already penetrated the cells and is now invisible, thus only effective in HPV-naive individuals. In addition, the cost and cold chain requirement renders it difficult to ensure wide-scale deployment to developing countries. Information about some prophylactic HPV vaccine clinical trials was reported in a previous study by the authors (12).
The limitations associated with prophylactic HPV vaccines resulted in the need to develop therapeutic HPV vaccines. These vaccines are geared towards preventing the progression of low-grade lesions, facilitating regression of existing lesions, controlling the spread of metastatic cancer, and preventing cancer recurrence post-treatment. This can be achieved by eliciting a cell-mediated immune response towards the intercellular virus. Several researchers have argued on the best target for therapeutic HPV vaccines, with some suggesting E1 and E2, some suggesting E5, and some suggesting E6 and E7. However, the latter is expressed in an advanced stage of the disease and is absent in normal cells, E5 promotes hyper-proliferation of infected cells in association with E6 and E7, and E1 and E2 are vital for viral replication, expressed in higher amounts and at an early stage of infection than E6 and E7. The outcome of various ongoing clinical trials will reveal the optimal target for therapeutic HPV vaccines. Strategies for therapeutic HPV vaccines include bacterial vectors, viral vectors, peptides, protein-based, DNA-based, RNA-based, tumor-based and dendritic cell-based vaccines.
HPV vaccine clinical trials are ongoing for ~20 years, with the efficacy of the vaccines expected to be known in 2023. No outcome has yet been published possibly due to the COVID-19 pandemic and other factors that might have stalled the studies, but there is high optimism that therapeutic HPV vaccines will surface shortly. A cross sectional analysis of HPV vaccine clinical trials captured in the International Clinical Trials Registry platform (ICTRP) reported that 437 HPV vaccine clinical trials were registered between 1999 and 2022, majority (37.3%) were in phase 3, 14.2% were in phase 4, 14.0% were in phase 2, and 17% did not indicate the phase of the trial. Among these, 46.7% of the trials were studying immune responses to HPV vaccines, 19.9% were studying HPV vaccine efficacy, 17.8% were studying HPV vaccine safety, 5% were studying safety and immunogenicity of the vaccines, while 3.9% had an unclear primary outcome (53). Information about some therapeutic HPV vaccine clinical trials can be found in our previous study (12).
Recommendations for vaccination
Despite the lack of availability of all the three approved prophylactic HPV vaccines worldwide, it important to state that Gardasil 9 would provide the highest protection against cervical cancer, and other HPV-associated cancers, as it targets more variants of HPV. However, the vaccination approach differs according to age, sex and risk of exposure (54). For individuals ≤26 years of age, HPV vaccines should be administered at 11 to 12 years of age, and for adolescents up to 26 years of age who have not been previously vaccinated or failed to complete the dosage, a catch-up vaccination is recommended according to the Advisory Committee on Immunization Practices (ACIP). For individuals aged >26 years, a catch-up vaccine is recommended (Grade 2C-very weak recommendation due to low quality evidence from observational studies, randomized clinical trials with serious flaws, or unsystematic clinical observations, according to the ACIP). This is due to the fact that the majority of individuals in this age category have been exposed to HPV; therefore, vaccination is not likely to improve immunity. However, some exceptions exists for healthcare workers and previously unvaccinated adults with little/no prior sexual experience (55).
8. Oncogenomics of cervical cancer
It was previously believed that only genetic alterations support HPV in cervical cancer pathogenesis; however, recent findings have indicated the crucial role of epigenetics in cervical cancer initiation and progression (56-59). Different epigenetic modifications, such as DNA methylation, histone modification and apoptotic regulation by non-coding RNAs in both the viral and host genome have been observed.
The majority of mammalian genomes are naturally methylated; however, the methylation of the promoter and its consecutive heterochromatic regions downregulates gene expression. The DNA methylation of tumor suppressor genes (TSGs) or oncogenes however, results in the expression of that gene. Thus, when a TSG is hypermethylated, it becomes silenced, leading to cellular transformation, genetic instability and cancer development. DNA methylation is a reversible epigenetic process that can be used to develop epigenetic markers for early diagnosis of cervical cancer. Genes such as RASSF1A and DAPK1 are associated with cervical cancer initiation and are hypermethylated in patients with cervical cancer (27).
For HPV on the other hand, its hypermethylation suppresses cellular carcinogenic transformation, while its hypomethylation leads to malignancy. The HPV oncogenes E5, E6, and E7 are naturally carcinogenic and their expression is controlled by the p97 promoter on the LCR. The E5 protein is found in the plasma membrane and Golgi apparatus of infected cells. Due to its weak oncogenic property, E5 inhibits the expression of MHC class 1 and increases the expression of epidermal growth factor receptor, thereby regulating the MAPK pathway. The E5 protein of HPV 16 is a fusogenic protein that localizes plasma membrane, is highly hydrophobic and induces cell-to-cell fusion leading to the formation of tetraploid cells. Such tetraploid cells do not undergo normal mitosis, resulting in apoptosis and p53-dependent cell cycle arrest (16,27). Aneuploidy and chromosomal instability facilitate HPV genome integration, leading to enhanced expression of E6 and E7 genes thereby supporting the growth of transformed cells. Furthermore, HR-HPV E5-induced cell fusion is a crucial event in the early stage of HPV-induced cervical cancer (60).
The E6 protein binds to several cellular factors, among which are p53, p300, myc, autocrine motility factor 1, interferon regulatory factor 3 (IRF3), pro-apoptotic Bc12 and BAK and E6 target protein 1 (E6TP1) among others. During infection, E6 causes the degradation of p53 by forming a complex with E3 ubiquitin ligase, inhibits the binding of p53 to its specific DNA sequence, sequestrates p53 in the cytoplasm hence inhibiting p53 signaling and finally abrogates the transactivation of p53 responsive gene, hence inhibiting apoptotic signal responsible for eliminating infected cells (16). Therefore, E6 regulates transcription and host cell signaling. Concerning innate immune response to viral infection, E6 interacts with Toll-like receptor 9 and IRF-3. Viral infection activates IRF-3, leading to the transcription of IFN-β, while the activation of Toll-like receptor 9 results in the production of cytokines, hence defending the cell from invading virus (61). E6 facilitates the development of cervical cancer by activating telomerase reverse transcriptase (hTERT) by inducing histone acetylation at the hTERT promoter. It also interacts with proteins involved in maintaining the chromosomal stability of infected cells, such as those involved in DNA break repair. E6-mediated p53 loss leads to the inactivation of G1 and G22 checkpoints giving rise to aneuploidy, genomic instability and progression to cancer. Actin fibers and normal cell structure are disrupted by the interaction of E6 and adhesion molecules that joins the cytoskeleton to the extracellular matrix (62). The anoikis apoptotic pathway is also targeted by E6, hence to some extent immortalizing HPV-infected cells (63).
The E7 protein of HR-HPV assists in cell cycle deregulation by the action of cyclin-dependent kinases (CDKs). It degrades pRB by proteasomal degradation and also activates E2F-mediated transcription, hence leading to S-phase competence in differentiated epithelial cells. However, the E7 protein of HPV 16 has been found to annul the inhibitory activities of cyclin-dependent kinase inhibitors. Therefore this function, in addition to the degradation of the pRB/E2F complex, results in maintaining differentiating keratinocytes in a competent state (16,64). The mechanism of cell death induced by HPV16 E7 protein is dependent upon the integrity of the p53 tumor suppressor pathway and appears to be different from the classical apoptosis because cell death is caspase-independent. HPV E7 protein is accompanied by a 600-kDa retinoblastoma factor (p600), and their interaction inhibits anoikis in detached cells, leading to increased viral transformation in such cells (63,65). E2F1, which mediates transcriptional control of the E2F6 gene binds to HPV16 E7. This interaction exerts a negative effect on E2F promoter activities, leading to differentiation and cell cycle exit. However, the direct interaction of HPV E7 with E2F6 leads to the functional deregulation of E2F6 and hence results in maintaining cells in an S-phase-competent state, thereby ensuring the continuation of the viral cycle (66). HPV16 E7-expressing cells have DNA repair foci which interfere with DNA break repair thereby enhancing viral genome integration. DNA double-stranded break results in the breaking off and integration of double-stranded HPV DNA into the host genome, hence leading to an onion-skin replication pattern in the presence of E1 and E2 expression. This is evident in patients with Fanconi anemia who have been found to have an increased incidence of cancer than others due to the increased ability of HPV16 E7 to induce DNA repair foci (67).
The tails of histone proteins protrude from nucleosomes (structural units of chromatin composed of DNA and five histones) providing access to post-translational modifications, with different types coexisting in the same tail and having a distinct effect. The regulation of histone acetylation is achieved by the action of histone acetyltransferases and histone deacetylases (HDACs), which regulate gene transcription by modifying the spatial structure of the nucleosome. The HDACs also known as eraser enzymes contains Rb ap48, a modulator that is essential in HPV16-induced cervical cancer transformation. The anticancer property of any molecule is affected by the degree of deacetylation. Therefore, HDAC inhibitor decreases histone deacetylation and increases histone acetylation, hence regulating gene expression, and leading to the build-up of reactive oxygen species and apoptosis (27).
Non-coding RNAs (ncRNAs) are essential biological regulators that control homeostasis and constitute ~70% of the human genome. Three non-coding RNAs, microRNAs (miRNAs), circular RNAs (circRNAs) and long non-coding RNAs (lncRNAs) are expressed in cancer progression and target DNA repair mechanisms, apoptosis and cell proliferation (27). In cervical cancer, miRNAs related to apoptosis are deregulated and function as either tumor suppressor genes or oncogenes. miR-744 enhances apoptosis and inhibits tumor growth by regulating Bcl-2(68). Likewise, miR-148b induces caspase-3-dependent apoptosis, and arrest cell cycle at G1/S by regulating cyclin D1 expression (69). In addition, miR-125b inhibits tumor growth and promotes apoptosis by targeting phosphoinositide 3-kinase catalytic subunit delta, while miR-181a causes resistance to radiation therapy by inhibiting Protein kinase C δ polypeptide chain, a pro-apoptotic gene (70). The lncRNAs have a size greater than 200 bp and lack ORFs, and hence are unable to code for proteins; however, they are essential in gene expression, cell cycle, cell differentiation and apoptosis. In cervical cancer, lncRNAs contribute significantly in cancer initiation and progression, inhibit apoptosis and promote cell growth via the mTOR pathway (27).
9. Conclusion and future prospects
The present review aimed to elucidate the history, epidemiology, etiology, risk factors, pathogenesis, oncogenomics, diagnosis and treatment of cervical cancer. Decades of research on the association between HPV and cervical cancer has shown that with the available information and tools at hand, it is possible to prevent most, if not all cases of cervical cancer. Cervical cancer is the fourth most common type of cancer, and the second most common type of cancer among women worldwide. In addition, with the rising incidence of infection annually, in addition to limited coverage rendered by the available prophylactic HPV vaccines, it is paramount to ensure further molecular studies towards understanding the genomics, epigenomics, transcriptomics implicated in cervical cancer, and to push towards completing the ongoing clinical trials on therapeutic HPV vaccines that aim to prevent progression of low-grade lesions, facilitate regression of existing lesions, control the spread of metastatic cancer, and prevent cancer recurrence post-treatment. To ensure a reliable decrease in incidence of cervical cancer, there is need for the governments, health agencies and all stakeholders work together in ensuring the incorporation of HPV testing into screening algorithms, especially for high risk groups, and ensuring an effective vaccine coverage for all adolescents. Equally, the development of robust population management systems is warranted to ensure the monitoring of vaccination, and the tracking of patients for a successful implementation of screenings and interventions.
Looking forward, advanced effective treatment strategies, such as immunotherapy and therapeutic HPV vaccines provide a promising avenue that will ensure a significant decrease in the incidence of cervical cancer, while controlling established infections or diseases. Furthermore, understanding the epigenetic markers and molecular mechanisms involved in cervical cancer will provide an avenue for novel diagnostic and personalized treatment approaches. The incorporation of computational approaches, molecular docking and simulations, and artificial intelligence is essential in the quest for improved and rapid screening approaches that can detect the slightest possible indication of infection/disease, as well as identify innovative cost-effective treatment strategies.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
IMK was involved in the literature search, in the writing of the manuscript and in the preparation of the figures. RIMDAR reviewed the manuscript. Both authors have read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
References
|
Buskwofie A, David-West G and Clare CA: A review of cervical cancer: Incidence and disparities. J Natl Med Assoc. 112:229–232. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Malik KI, Aliyu DU, Abubakar BJ, Lukman Y, Sale KA, Alkali BH, Saidu A, Bala DA, Umoru A, Lawal N and Abubakar AI: Identification of high-risk human papillomavirus isolates circulating in Nigeria and phylogenetic analysis based on the virus essential protein. Indian J Gynecol Oncol. 19(87)2021. | |
|
Singh D, Vignat J, Lorenzoni V, Eslahi M, Ginsburg O, Lauby-Secretan B, Arbyn M, Basu P, Bray F and Vaccarella S: Global estimates of incidence and mortality of cervical cancer in 2020: A baseline analysis of the WHO global cervical cancer elimination initiative. Lancet Glob Heal. 11:e197–e206. 2023.PubMed/NCBI View Article : Google Scholar | |
|
Vu M, Yu J, Awolude OA and Chuang L: Cervical cancer worldwide. Curr Probl Cancer. 42:457–465. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Siegel RL, Miller KD and Jemal A: Cancer statistics, 2020. CA Cancer J Clin. 70:7–30. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Collins Y, Holcomb K, Chapman-Davis E, Khabele D and Farley JH: Gynecologic cancer disparities: A report from the health disparities taskforce of the society of gynecologic oncology. Gynecol Oncol. 133:353–361. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Siegel RL, Kratzer TB, Giaquinto AN, Sung H and Jemal A: Cancer statistics, 2025. CA Cancer J Clin. 75:10–45. 2025.PubMed/NCBI View Article : Google Scholar | |
|
Wu J, Jin Q, Zhang Y, Ji J, Li J, Liu X, Duan H, Feng Z, Liu Y, Zhang Y, et al: Global burden of cervical cancer: Current estimates, temporal trend and future projections based on the GLOBOCAN 2022. J Natl Cancer Cent. 23:1–24. 2025. | |
|
zur Hausen H: Papillomaviruses and cancer: From basic studies to clinical application. Nat Rev Cancer. 2:342–350. 2002.PubMed/NCBI View Article : Google Scholar | |
|
Akinlotan M, Bolin JN, Helduser J, Ojinnaka C, Lichorad A and McClellan D: Cervical cancer screening barriers and risk factor knowledge among uninsured women. J Community Health. 42:770–778. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Johnson CA, James D, Marzan A and Armaos M: Cervical cancer: An overview of pathophysiology and management. Semin Oncol Nurs. 35:166–174. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Kabir IM, Dutsinma UA, Bala JA, Yusuf L, Abubakar SD, Kumurya AS, Bulama HA, Bello ZM and Aliyu IA: The need for therapeutic hpv vaccines as a means of curbing the menace of cervical cancer. Indian J Gynecol Oncol. 19(96)2021. | |
|
Yousefi Z, Aria H, Ghaedrahmati F, Bakhtiari T, Azizi M, Bastan R, Hosseini R and Eskandari N: An update on human papilloma virus vaccines: History, types, protection, and efficacy. Front Immunol. 12(805695)2022.PubMed/NCBI View Article : Google Scholar | |
|
Jain MA and Limaiem F: Human Papillomavirus and Related Diseases From Bench to Bedside A Diagnostic and Preventive Perspective. Treasure Island, FL, 2023. | |
|
Broeck DV: Human papillomavirus and related diseases-from bench to bedside. Broeck DV (ed). IntechOpen, p336, 2012. | |
|
Herfs M, Hubert P, Moutschen M and Delvenne P: Mucosal junctions: Open doors to HPV and HIV infections? Trends Microbiol. 19:114–120. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Yuan F, Auborn K and James C: Altered growth and viral gene expression in human papillomavirus type 16-containing cancer cell lines treated with progesterone. Cancer Invest. 17:19–29. 1999.PubMed/NCBI | |
|
Jing Y, Wang T, Chen Z, Ding X, Xu J, Mu X, Cao M and Chen H: Phylogeny and polymorphism in the long control regions E6, E7, and L1 of HPV type 56 in women from Southwest China. Mol Med Rep. 17:7131–7141. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Kuguyo O, Tsikai N, Thomford NE, Magwali T, Madziyire MG, Nhachi CFB, Matimba A and Dandara C: Genetic susceptibility for cervical cancer in African populations: What are the host genetic drivers? OMICS. 22:468–483. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Zhang Y, Fan S, Meng Q, Ma Y, Katiyar P, Schlegel R and Rosen EM: BRCA1 interaction with human papillomavirus oncoproteins. J Biol Chem. 280:33165–33177. 2005.PubMed/NCBI View Article : Google Scholar | |
|
Zhou X, Han S, Wang S, Chen X, Dong J, Shi X, Xia Y, Wang X, Hu Z and Shen H: Polymorphisms in HPV E6/E7 protein interacted genes and risk of cervical cancer in Chinese women: A case-control analysis. Gynecol Oncol. 114:327–331. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Bodily J and Laimins AL: Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 19:33–39. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Baseman JG and Koutsky LA: The epidemiology of human papillomavirus infections. J Clin Virol. 32:16–24. 2005.PubMed/NCBI View Article : Google Scholar | |
|
Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M and Huh K: Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 78:11451–11460. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Schiffman M and Wentzensen N: Human papillomavirus (HPV) infection and the multi-stage carcinogenesis of cervical cancer introduction and historic context. Cancer Epidemiol Biomarkers Prev. 22:553–560. 2013. | |
|
Yadav C, Yadav R, Chabbra R, Nanda S, Ranga S, Kadian L and Ahuja P: Overview of genetic and epigenetic regulation of human papillomavirus and apoptosis in cervical cancer. Apoptosis. 28:683–701. 2023.PubMed/NCBI View Article : Google Scholar | |
|
Smola S, Trimble C and Stern PL: Human papillomavirus-driven immune deviation: Challenge and novel opportunity for immunotherapy. Ther Adv Vaccines. 5:69–82. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Gutiérrez-Hoya A and Soto-Cruz I: NK cell regulation in cervical cancer and strategies for immunotherapy. Cells. 10(3104)2021.PubMed/NCBI View Article : Google Scholar | |
|
Tamoutounour S, Han SJ, Deckers J, Constantinides MG, Hurabielle C, Harrison OJ, Bouladoux N, Linehan JL, Link VM, Vujkovic-Cvijin I, et al: Keratinocyte-intrinsic MHCII expression controls microbiota-induced Th1 cell responses. Proc Natl Acad Sci USA. 116:23643–23652. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Manzo-Merino J, Del-Toro-Arreola S, Rocha-Zavaleta L, Peralta-Zaragoza Ó, Jiménez-Lima R and Madrid-Marina V: Immunology of cervical cancer. Rev Invest Clin. 72:188–197. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Stanley MA: Immune responses to human papilloma viruses. Indian J Med Res. 130:266–276. 2009.PubMed/NCBI | |
|
Stanley M: Immunobiology of HPV and HPV vaccines. Gynecol Oncol. 109 (Suppl 2):S15–S21. 2008.PubMed/NCBI View Article : Google Scholar | |
|
Tindle RW: Cervical Cancer. StatPearls Publishing, Treasure Island, FL, pp1-7, 2002. | |
|
Doorbar J, Egawa N, Griffin H, Kranjec C and Murakami I: Human papillomavirus molecular biology and disease association. Rev Med Virol. 25 (Suppl 1):S2–S23. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Siddiqa A, Sims-Mourtada JC, Guzman-Rojas L, Rangel R, Guret C, Madrid-Marina V, Sun Y and Martinez-Valdez H: Regulation of CD40 and CD40 ligand by the AT-hook transcription factor AKNA. Nature. 410:383–387. 2001.PubMed/NCBI View Article : Google Scholar | |
|
Garcia-Iglesias T, Del Toro-Arreola A, Albarran-Somoza B, Del Toro-Arreola S, Sanchez-Hernandez PE, Ramirez-Dueñas MG, Balderas-Peña LM, Bravo-Cuellar A, Ortiz-Lazareno PC and Daneri-Navarro A: Low NKp30, NKp46 and NKG2D expression and reduced cytotoxic activity on NK cells in cervical cancer and precursor lesions. BMC Cancer. 9(186)2009.PubMed/NCBI View Article : Google Scholar | |
|
Sasson IM, Haley NJ, Hoffmann D, Wynder EL, Hellberg D and Nilsson S: Cigarette smoking and neoplasia of the uterine cervix: Smoke constituents in cervical mucus. N Engl J Med. 312:315–316. 1985.PubMed/NCBI View Article : Google Scholar | |
|
Roteli-Martins CM, Panetta K, Alves VA, Siqueira SA, Syrjänen KJ and Derchain SF: Cigarette smoking and high-risk HPV DNA as predisposing factors for high-grade cervical intraepithelial neoplasia (CIN) in young Brazilian women. Acta Obstet Gynecol Scand. 77:678–682. 1998.PubMed/NCBI View Article : Google Scholar | |
|
Stumbar SE, Stevens M and Feld Z: Cervical cancer and its precursors: A preventative approach to screening, diagnosis, and management. Prim Care. 46:117–134. 2019.PubMed/NCBI View Article : Google Scholar | |
|
International Collaboration of Epidemiological Studies of Cervical Cancer. Comparison of risk factors for invasive squamous cell carcinoma and adenocarcinoma of the cervix: Collaborative reanalysis of individual data on 8,097 women with squamous cell carcinoma and 1,374 women with adenocarcinoma from 12 epidemiological studies. Int J Cancer. 120:885–891. 2007.PubMed/NCBI View Article : Google Scholar | |
|
Burd EM: Human papillomavirus and cervical cancer. Clin Microbiol Rev. 16:1–17. 2003.PubMed/NCBI View Article : Google Scholar | |
|
Olusola P, Banerjee HN, Philley JV and Dasgupta S: Human papilloma virus-associated cervical cancer and health disparities. Cells. 8(622)2019.PubMed/NCBI View Article : Google Scholar | |
|
Fowler JR, Maani EV, Dunton CJ and Jack BW: Cervical Cancer. StatPearls Publishing, Treasure Island, FL, 2023. | |
|
Burness JV, Schroeder JM and Warren JB: Cervical colposcopy: Indications and risk assessment. Am Fam Physician. 102:39–48. 2020.PubMed/NCBI | |
|
Sachan P, Singh M, Patel M and Sachan R: A study on cervical cancer screening using pap smear test and clinical correlation. Asia Pac J Oncol Nurs. 5:337–341. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Turinetto M, Valsecchi AA, Tuninetti V, Scotto G, Borella F and Valabrega G: Immunotherapy for cervical cancer: Are we ready for prime time? Int J Mol Sci. 23(3559)2022.PubMed/NCBI View Article : Google Scholar | |
|
Lazo PA: The molecular genetics of cervical carcinoma. Br J Cancer. 80:2008–2018. 1999.PubMed/NCBI View Article : Google Scholar | |
|
Mezache L, Paniccia B, Nyinawabera A and Nuovo GJ: Enhanced expression of PD L1 in cervical intraepithelial neoplasia and cervical cancers. Mod Pathol. 28:1594–1602. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK and Sharpe AH: PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 206:3015–3029. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Jenkins RW, Barbie DA and Flaherty KT: Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 118:9–16. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Kakotkin VV, Semina EV, Zadorkina TG and Agapov MA: Prevention strategies and early diagnosis of cervical cancer: Current state and prospects. Diagnostics (Basel). 13(610)2023.PubMed/NCBI View Article : Google Scholar | |
|
Mavundza EJ, Mmotsa TM and Ndwandwe D: Human papillomavirus (HPV) trials: A cross-sectional analysis of clinical trials registries. Hum Vaccin Immunother. 20(2393481)2024.PubMed/NCBI View Article : Google Scholar | |
|
Saslow D, Andrews KS, Manassaram-Baptiste D, Smith RA and Fontham ETH: American Cancer Society Guideline Development Group. Human papillomavirus vaccination 2020 guideline update: American cancer society guideline adaptation. CA Cancer J Clin. 70:274–280. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Eun TJ and Perkins RB: Screening for cervical cancer. Med Clin North Am. 104:1063–1078. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Pardini B, De Maria D, Francavilla A, Di Gaetano C, Ronco G and Naccarati A: MicroRNAs as markers of progression in cervical cancer: A systematic review. BMC Cancer. 18(696)2018.PubMed/NCBI View Article : Google Scholar | |
|
Gu Y, Feng X, Jin Y, Liu Y, Zeng L, Zhou D and Feng Y: Upregulation of miRNA-10a-5p promotes tumor progression in cervical cancer by suppressing UBE2I signaling. J Obstet Gynaecol. 43(2171283)2023.PubMed/NCBI View Article : Google Scholar | |
|
Lev Maor G, Yearim A and Ast G: The alternative role of DNA methylation in splicing regulation. Trends Genet. 31:274–280. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Boland CR: Non-coding RNA: It's not junk. Dig Dis Sci. 62:1107–1109. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Gao P and Zheng J: High-risk HPV E5-induced cell fusion: A critical initiating event in the early stage of HPV-associated cervical cancer. Virol J. 7(238)2010.PubMed/NCBI View Article : Google Scholar | |
|
Hasan UA, Bates E, Takeshita F, Biliato A, Accardi R, Bouvard V, Mansour M, Vincent I, Gissmann L, Iftner T, et al: TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J Immunol. 178:3186–3197. 2007.PubMed/NCBI View Article : Google Scholar | |
|
Howie HL, Katzenellenbogen RA and Galloway DA: Papillomavirus E6 proteins. Bone. 23:1–7. 2008.PubMed/NCBI View Article : Google Scholar | |
|
Huh KW, DeMasi J, Ogawa H, Nakatani Y, Howley PM and Münger K: Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc Natl Acad Sci USA. 102:11492–11497. 2005.PubMed/NCBI View Article : Google Scholar | |
|
Zerfass K, Schulze A, Spitkovsky D, Friedman V, Henglein B and Jansen-Dürr P: Sequential activation of cyclin E and cyclin A gene expression by human papillomavirus type 16 E7 through sequences necessary for transformation. J Virol. 69:6389–6399. 1995.PubMed/NCBI View Article : Google Scholar | |
|
Eichten A, Rud DS, Grace M, Piboonniyom SO, Zacny V and Münger K: Molecular pathways executing the ‘trophic sentinel’ response in HPV-16 E7-expressing normal human diploid fibroblasts upon growth factor deprivation. Virology. 319:81–93. 2004.PubMed/NCBI View Article : Google Scholar | |
|
McLaughlin-Daurbin ME: Oncogenic activities of human papillomaviruses margaret. Virus Res. 71:3831–3840. 2014. | |
|
Spardy N, Duensing A, Charles D, Haines N, Nakahara T, Lambert PF and Duensing S: The human papillomavirus type 16 E7 oncoprotein activates the fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J Virol. 81:13265–13270. 2007.PubMed/NCBI View Article : Google Scholar | |
|
Chen XF and Liu Y: MicroRNA-744 inhibited cervical cancer growth and progression through apoptosis induction by regulating Bcl-2. Biomed Pharmacother. 81:379–387. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Mou Z, Xu X, Dong M and Xu J: MicroRNA-148b acts as a tumor suppressor in cervical cancer by inducing G1/S-Phase cell cycle arrest and apoptosis in a caspase-3-dependent manner. Med Sci Monit. 22:2809–2815. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Liu S, Zhang P, Chen Z, Liu M, Li X and Tang H: MicroRNA-7 downregulates XIAP expression to suppress cell growth and promote apoptosis in cervical cancer cells. FEBS Lett. 587:2247–2253. 2013.PubMed/NCBI View Article : Google Scholar |









