



Histone deacetylase 6: A new player in oxidative stress‑associated disorders and cancers (Review)
- Authors:
- Published online on: July 4, 2025 https://doi.org/10.3892/ijmm.2025.5578
- Article Number: 137
-
Copyright: © Qu et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
The development of numerous long-term illnesses is caused by oxidative stress, which occurs when the body cannot effectively neutralize the damaging effects of reactive oxygen species (ROS) due to an imbalance in production and detoxification processes (1). This lack of balance results in cellular damage, contributing to the development of various illnesses, such as neurodegenerative diseases, cancer, heart diseases and metabolic disorders (2-5). The cellular response to oxidative stress involves a complex network of signaling pathways that orchestrate defense mechanisms, repair processes and at times cell death (6). Among the myriad regulators of these pathways, histone deacetylase 6 (HDAC6) emerges as a pivotal player due to its unique functions and substrates. HDAC6, part of the class IIb HDAC group, is mainly found in the cytoplasm and has distinct characteristics that set it apart from other HDACs, which mainly reside in the nucleus and control gene expression through histone deacetylation (7). HDAC6 is known for removing acetyl groups from non-histone proteins such as α-tubulin, cortactin and heat shock protein 90 (HSP90), which play vital roles in preserving cell structure, movement and response to stress (8-10). Through its influence on these substrates, HDAC6 directly impacts cellular dynamics, signaling pathways and survival mechanisms under oxidative stress conditions.
The role of HDAC6 in oxidative stress is paradoxical and multifaceted. On the one hand, HDAC6 promotes cellular survival by maintaining the dynamic stability of the cytoskeleton through the deacetylation of α-tubulin (11). This action facilitates the proper assembly of microtubules, essential for cell shape, intracellular transport and the initiation of cell division. On the other hand, HDAC6 can exacerbate oxidative stress by influencing other pathways. For instance, HDAC6 affects the acetylation status of cortactin, a promoter of actin polymerization, thus modulating the cell's ability to form stress fibers and lamellipodia in response to external stress signals (12). In addition, HDAC6 regulates the function of HSP90, a chaperone that stabilizes several client proteins, including those involved in cell growth and survival signaling (13). The deacetylation of HSP90 by HDAC6 enhances its chaperone activity, thereby increasing the stability and function of its client proteins, many of which are critical in the response to oxidative stress. This modulation can have protective roles in cells by maintaining protein homeostasis and function during stress conditions.
The involvement of HDAC6 in disease is linked to its roles in modulating oxidative stress and cellular homeostasis. Neurodegenerative disorders like Alzheimer's and Parkinson's can be exacerbated by excessive acetylation caused by the overactive HDAC6 enzyme, leading to the accumulation of improperly folded proteins, a key feature of these conditions (14,15). By contrast, inhibiting HDAC6 has shown potential in reducing these aggregates, suggesting a therapeutic angle for modulating HDAC6 activity. In cancer, HDAC6 plays a role in cellular transformation and malignancy through its effects on the acetylation of oncogenes and tumor suppressor genes. Its activity influences cancer cell migration, invasion and resistance to chemotherapeutic agents, primarily through its regulation of the cytoskeleton and cell adhesion molecules (16). HDAC6 also regulates how heart and blood vessel cells respond to oxidative stress in cardiovascular conditions, potentially causing heart attacks, high blood pressure and hardening of the arteries (17,18). The regulation of HDAC6 activity has been shown to affect the heart's response to stress by modulating inflammatory responses and myocardial contraction (19).
Given its central role in multiple disease-related pathways, HDAC6 represents a promising target for therapeutic intervention. The selective inhibition of HDAC6 offers a way to modulate its activity in disease without affecting the global acetylation levels that could result from broadly inhibiting HDACs. Various inhibitors targeting HDAC6, including ricolinostat, tubastatin A (TubA) and ACY-1215, have been developed and are undergoing clinical trials to evaluate their effectiveness in treating cancer and neurodegenerative disorders (19-21). These inhibitors have shown promise in preclinical models, reducing disease pathology and improving survival. Furthermore, beyond pharmacological inhibitors, understanding the regulation of HDAC6 activity at the transcriptional, post-transcriptional and post-translational levels provides additional layers of potential intervention. For instance, targeting the upstream kinases that phosphorylate HDAC6, thereby modulating its activity, or manipulating its protein-protein interactions, could offer new ways to fine-tune HDAC6's functions specifically in oxidative stress-related pathologies.
Investigating HDAC6 in relation to oxidative stress provides new opportunities for understanding its dual roles in cell viability and apoptosis. Its broad impact on disease through the modulation of protein acetylation provides both challenges and opportunities for developing targeted therapies. As research continues to unravel the complexities of HDAC6 function, the potential to harness its activity for therapeutic benefit in oxidative stress-related diseases becomes increasingly feasible. The purpose of this review is to offer a thorough examination of the existing knowledge on HDAC6, emphasizing its significance in maintaining cellular balance and exploring its potential in treating diseases through targeted therapies.
HDAC6: An overview
HDAC6, a key player in the HDAC family, is known for its significant involvement in a range of cellular functions, particularly those related to protein breakdown, cell movement and response to stress (7). HDAC6 stands out within the HDAC family because of its large size and possession of two separate catalytic domains, CD1 and CD2, in addition to a zinc-finger domain (ZnF-UBP) that binds ubiquitin at the end of the protein (22).
Structure of HDAC6
HDAC6, composed of 1,215 amino acids, stands as the largest HDAC. Primarily located in the cytoplasm, this positioning is influenced by the SE14 motif (a tetra-decapeptide repeat domain containing Glu-Ser) and two conserved nuclear export signals (NES1 and NES2) in the N-terminal region (23). The C-terminal ZnF-UBP is essential for binding polyubiquitinated proteins, aiding their degradation through the aggresome-autophagy pathway. This domain enables HDAC6 to interact with ubiquitinated misfolded proteins, directing them to aggresomes for autophagic degradation, thus maintaining cellular protein quality control (24). The two catalytic domains of HDAC6, CD1 and CD2, although similar in structure, have distinct functional roles. CD1 has a broader active site but more limited substrate specificity, preferring peptide substrates with C-terminal acetyllysine residues. On the other hand, CD2 has a greater impact on the total catalytic function of HDAC6, particularly in the deacetylation of tubulin and tau proteins (25). This functional differentiation is crucial for the enzyme's involvement in microtubule dynamics and cell motility.
The structure of HDAC6 is composed of several domains that confer its multifunctional nature. The enzyme's N-terminal region includes a dynein motor-binding domain that assists in the transportation of misfolded proteins to aggresomes along microtubules by HDAC6 (26). This domain interacts with the dynein motor complex, which is essential for retrograde transport within the cell. The linker region connecting the catalytic domains CD1 and CD2 can impact the spatial orientation and availability of substrates. The C-terminal ZnF-UBP domain binds ubiquitinated proteins, linking HDAC6 to the ubiquitin-proteasome system (27).
Function of HDAC6
HDAC6 plays a multifaceted role in cellular homeostasis, extending beyond its traditional function of histone deacetylation. Non-histone proteins, such as α-tubulin, HSP90 and cortactin, are the main targets of this enzyme, playing crucial roles in cell movement, protein breakdown and response to stress.
Microtubule dynamics and cell motility
HDAC6 deacetylates α-tubulin, a key component of the microtubule network. This deacetylation reduces the stability of microtubules, promoting increased cell motility and invasiveness, characteristics often exploited by cancer cells (28). Inhibition of HDAC6 leads to hyperacetylation of α-tubulin, resulting in stabilized microtubules and decreased cell motility (11). This function plays a vital role in activities such as cell movement, axonal transport and intracellular substance movement. Additionally, HDAC6 interacts with cortactin, a protein that binds to actin and controls the movement of the actin cytoskeleton. Cortactin is involved in the formation of lamellipodia and invadopodia, structures essential for cell movement and invasion (29). HDAC6-mediated deacetylation of cortactin enhances its ability to bind to F-actin, thereby promoting actin polymerization and cell motility.
Protein degradation
HDAC6 interacts with HSP90, a molecular chaperone essential for the stability and function of numerous client proteins involved in cell growth and survival (30). By deacetylating HSP90, HDAC6 enhances its chaperone activity, which is crucial for the proper folding of nascent proteins and the degradation of misfolded proteins (30). This interaction is particularly important in cells under stress, where the accumulation of misfolded proteins can lead to cellular dysfunction and disease (30). One of the key roles of HDAC6 is in the aggresome-autophagy pathway (31). When the ubiquitin-proteasome system is overwhelmed or impaired, misfolded and aggregated proteins are transported to aggresomes, which are perinuclear inclusion bodies (31). HDAC6 facilitates the transport of these ubiquitinated proteins along microtubules to aggresomes by binding to both the ubiquitinated cargo and the dynein motor complex. Once sequestered in aggresomes, these proteins can be degraded by autophagy, a lysosome-dependent degradation pathway (31). This process is essential for maintaining protein homeostasis and preventing the accumulation of toxic protein aggregates.
Aggresome formation and autophagy
HDAC6 facilitates the formation of aggresomes, cellular structures that sequester misfolded proteins (32). By binding to ubiquitinated proteins through its ZnF-UBP domain, HDAC6 transports these proteins to the microtubule-organizing center, where they are sequestered into aggresomes and subsequently degraded by autophagy (27). This function is vital for cell survival under conditions of proteotoxic stress. The aggresome pathway is particularly important in neurons, which are highly susceptible to the accumulation of misfolded proteins due to their long lifespan and complex structure. Inhibition of HDAC6 has been shown to impair aggresome formation and autophagy, leading to increased sensitivity to proteotoxic stress and neuronal degeneration (33).
Oxidative stress response
HDAC6 influences the cellular response to oxidative stress through its impact on mitochondrial activity. It has been shown to affect mitochondrial dynamics and biogenesis through interactions with proteins involved in mitochondrial fusion and fission (34). Blocking HDAC6 activity may result in elevated generation of reactive oxygen species (ROS) and impairment of mitochondria, underscoring its importance in maintaining cellular redox balance (35). Mitochondria play a crucial role in generating ROS within cells, with the equilibrium between mitochondrial fusion and fission being essential for preserving mitochondrial function and preventing oxidative damage (36). HDAC6 interacts with dynamin-related protein 1, a crucial regulator of mitochondrial division, enhancing its deacetylation and activation. This interaction facilitates the division of damaged mitochondria and their removal by mitophagy, a selective form of autophagy that targets dysfunctional mitochondria (37).
HDAC6 is involved in the cellular stress response by regulating the acetylation status of HSPs. HSPs act as molecular chaperones, aiding in the folding of proteins and shielding cells from damage caused by stress. HDAC6 deacetylates HSP90, enhancing its chaperone activity and promoting the stabilization and refolding of stress-damaged proteins (38). This function is particularly important under conditions of oxidative stress, where the accumulation of damaged proteins can lead to cell death. In addition to HSP90, HDAC6 also deacetylates other stress-responsive proteins, such as HSP70 and HSP27 (39). These proteins play crucial roles in protecting cells from heat shock, oxidative stress and other environmental insults. By regulating the acetylation status of these proteins, HDAC6 modulates their chaperone activity and enhances the cellular capacity to cope with stress.
Regulation of signal transduction pathways
HDAC6 is essential for controlling various signal transduction pathways, such as NF-κB and MAPK, which are crucial for functions like inflammation, cell viability and programmed cell death (40,41). By deacetylating key components of these pathways, HDAC6 modulates their activity and impacts cellular responses to external stimuli. In the NF-κB pathway, HDAC6 deacetylates and stabilizes the p65 subunit, facilitating its nuclear translocation and transcriptional activity (42). This enhances the activation of NF-κB target genes related to inflammation and immune system function. Studies have shown that inhibiting HDAC6 can reduce NF-κB activity, thereby diminishing inflammatory responses in various cell types. In the MAPK pathway, HDAC6 interacts with and deacetylates crucial signaling molecules like ERK1/2 and JNK (43,44). These interactions impact the initiation and subsequent signaling of the MAPK pathway, which in turn affect cellular functions like growth, differentiation and programmed cell death. HDAC6 inhibition has been observed to alter MAPK signaling, thereby modulating cellular responses to stress and growth factors.
In summary, HDAC6 is integral to processes such as microtubule dynamics, protein degradation, aggresome formation, oxidative stress response, cellular stress response and signal transduction pathways, highlighting its crucial role in cell biology. Understanding the structure and function of HDAC6 provides important insights into its roles in health and disease, emphasizing its potential as a therapeutic target for various conditions. Continued investigation into how HDAC6 regulates these functions, as well as the development of specific HDAC6 inhibitors, holds promise for innovative treatment approaches. These strategies aim to modulate HDAC6 activity to treat diseases characterized by dysregulated protein homeostasis and stress responses.
Function of HDAC6 in oxidative stress-related disease
An imbalance between the production of ROS and the cell's capacity to counteract these reactive molecules or repair the consequent damage leads to oxidative stress. ROS are highly reactive molecules that can damage cellular components such as proteins, lipids and DNA, resulting in a range of health issues. HDAC6 plays a crucial role in regulating how cells respond to oxidative stress through its impact on mitochondrial activity and interaction with antioxidant systems. HDAC6 deacetylates non-histone proteins, affecting crucial cellular processes. Increased HDAC6 activity is associated with numerous pathological conditions where oxidative stress is a significant factor, such as neurodegenerative diseases, cardiovascular disorders, metabolic syndromes and cancers. HDAC6's regulation of mitochondrial function, protein degradation and stress responses highlights its importance in disease progression.
Function of HDAC6 in oxidative stress-induced neurodegenerative diseases
Cognitive impairments in individuals with obstructive sleep apnea (OSA) are associated with neuroinflammation and oxidative stress caused by intermittent hypoxia (45). New research has highlighted the involvement of TAR DNA-binding protein 43 (TDP-43), HDAC6 and peroxiredoxin 1 (Prdx1) in cognitive decline linked to various degenerative conditions. During this research, individuals with a diagnosis of OSA confirmed by polysomnography were evaluated for cognitive function using the Montreal Cognitive Assessment and blood samples were collected (46). TDP-43 and HDAC6 levels increased in HMC3 cells treated with lipopolysaccharide (LPS), whereas Prdx1 levels decreased. TDP-43 regulated Prdx1 expression by modulating HDAC6, with changes in inflammation and oxidative stress correlating with TDP-43 levels. Administering a specific HDAC6 inhibitor reduced inflammation and oxidative stress caused by LPS through the elevation of Prdx1 levels (46). The research findings suggest that TDP-43 plays a role in affecting Prdx1, leading to increased neuroinflammation and oxidative stress through the regulation of HDAC6 expression.
Parkinson's disease (PD) is a long-term degenerative condition marked by the death of dopamine-producing cells in the substantia nigra pars compacta and striatum, resulting in movement problems (47). Multiple pathogenic mechanisms contribute to PD, and currently, there is no cure. In the pathogenesis of PD, the inflammatory response is crucial, as postmortem examinations of PD patients' brains show increased levels of inflammatory cytokines, such as interleukin (IL)-1β and IL-18 (48). Interestingly, anti-inflammatory medications have demonstrated effectiveness in treating PD. Researchers investigated the impact of tubastatin A (TBA) on protein 3 containing NACHT, LRR and PYD structural domains (NLRP3) activation, SH-SY5Y cell damage and inflammatory response in a study utilizing a 6-hydroxydopamine (6-OHDA)-induced PD model. They also explored its effects on NLRP3 activation and dopaminergic damage in the nigrostriatal system of mice, assessing peroxiredoxin 2 (Prx2) acetylation levels and oxidative stress. The findings showed that TBA suppressed the activation of NLRP3 induced by 6-OHDA, leading to lower levels of NLRP3, mature caspase-1 and IL-1β, as well as reduced gliosis and degeneration of dopaminergic neurons (49). Significantly, TBA reinstated levels of Prx2 acetylation and decreased oxidative stress. Pharmacologically inhibiting HDAC6 using TBA appears to reduce NLRP3-induced inflammation and safeguard dopaminergic neurons, possibly by altering Prx2 acetylation (49). The research indicates that focusing on the deacetylase catalytic region of HDAC6 may offer a promising treatment approach for PD (Fig. 1).

The process of oxidation negatively impacts the ability to heal following a hemorrhage within the brain, and HDAC6 plays a crucial role in initiating this stress (50). Research involving HDAC6 knockout mice showed resistance to oxidative stress following intracerebral hemorrhage (ICH). Lack of HDAC6 resulted in decreased neuronal cell death and reduced levels of proteins associated with cell death. Studies on the mechanisms revealed that HDAC6 interacts with malate dehydrogenase 1 (MDH1) and removes acetyl groups from lysine residues 121 and 298. Exposure of HT22 cells to stressors related to ICH, such as hemoglobin and thrombin, resulted in the suppression of MDH1 acetylation, which was reversed upon HDAC6 inhibition, indicating a connection between HDAC6 and MDH1 (51). Acetylation-mimicking MDH1 mutants, unlike acetylation-resistant ones, protected neurons from oxidative damage. In addition, inhibiting HDAC6 did not decrease brain injury following ICH in the absence of MDH1 (51). Therefore, HDAC6 inhibition appears to mitigate brain damage during ICH by enhancing MDH1 acetylation (Fig. 1).
Most neurodegeneration data derive from acute toxin (e.g., 6-OHDA) or transgenic overexpression models that capture only single mechanisms of Parkinson's or Alzheimer's pathology (52,53). These paradigms fail to mimic the slow, multifactorial neuronal loss or the age- and sex-dependent penetrance observed in patients (52). HDAC6-knockout mice also show developmental compensation in microtubule dynamics, potentially masking late-life toxicities (54). Human data remain restricted to small post-mortem series; comorbidities and pharmacotherapy exposure confound HDAC6 expression estimates (55). Longitudinal biomarker-driven cohorts and patient-derived induced pluripotent stem cell models will be required to confirm disease stage-specific benefits or risks of HDAC6 inhibition (56,57).
Role of HDAC6 in oxidative stress-induced cardiovascular diseases
Heart failure involves gene expression regulated by HDACs, and inhibiting these enzymes has shown promise in treating this condition (17). In neonatal rat ventricular myocytes, researchers discovered that pretreatment with seleno-suberoylanilide hydroxamic acid (Se-SAHA) reduced cardiac hypertrophy and fibrosis induced by isoproterenol (ISO). In vitro, Se-SAHA markedly decreased the generation of ROS induced by ISO and restored the levels of superoxide dismutase (SOD)2 and heme oxygenase (HO)-1 expression. Furthermore, Se-SAHA pretreatment inhibited autophagosome buildup and reversed the upregulation of HDAC1 and HDAC6 caused by ISO exposure (58). In a mouse model induced by ISO, Se-SAHA alleviated ventricular systolic dysfunction, hypertrophy and fibrosis, while also suppressing the excessive expression of HDAC1 and HDAC6. Se-SAHA significantly boosted SOD2 activity, enhancing its ability to scavenge free radicals. In addition, Se-SAHA suppressed the increased amounts of microtubule-associated protein 1 light chain 3-II and Beclin-1 in mice with heart failure (58). In conclusion, Se-SAHA exerts cardioprotective effects on ISO-induced heart failure by reducing oxidative stress and inhibiting autophagy (Fig. 2).
Heart failure is often linked to telomere shortening in cardiomyocytes, with arterial hypertension being a primary risk factor (59). Both conditions involve dysregulation of the neurohormonal axis (60). Telomere length in cardiomyocytes was measured in a mouse model of hypertensive atrial fibrillation caused by increased neurohormonal activity [through angiotensin II (AngII) infusion, a high-salt diet and no nephrectomy], as well as in AngII-stimulated cardiomyocytes and endomyocardial biopsy tissues from patients with hypertensive atrial fibrillation. The findings showed that telomere shortening occurred in both laboratory settings and living organisms, associated with left ventricular enlargement and decreased left ventricular function. Telomere shortening was linked to higher levels of superoxide production, increased expression of NADPH oxidase 2 (NOX2), elevated activity of HDAC6, decreased levels of the telomere-specific antioxidant PRDX1 and increased oxidative DNA damage. Inhibition of NOX2 prevented the reduction of PRDX1, DNA damage and telomere shortening, emphasizing NOX2 as a critical producer of ROS (61). Combining the NOX inhibitor apocynin with other treatments reduced the effects of hypertensive hyperlipidemia and telomere shortening. Similarly, the HDAC6 inhibitor TubA, which increases PRDX1 availability, halted telomere shortening in mature heart muscle cells. Thus, the reduction in fractional blood loss caused by neurohormonal hyperactivation in patients with heart failure leads to telomere shortening and oxidative damage in cardiomyocytes, which is reliant on superoxide derived from NOX2 (61). These findings suggest potential targeted treatments for heart failure involving the inhibition of NOX2 and HDAC6 to mitigate oxidative stress and telomere shortening (Fig. 2).
According to the heart failure-gut theory, damage to the intestinal mucosa causes a rise in microbial translocation, resulting in changes to circulating metabolites that contribute to heart failure (62). In this research, human AC16 cells were exposed to doxorubicin to create a heart failure model in a laboratory setting. Scientists studied the impact of indole-3-propionic acid (IPA) on cell survival, programmed cell death, inflammatory response and oxidative damage. The findings indicated that IPA significantly reduced cell death, inflammation and oxidative damage in the treated cells. Structural analysis revealed that IPA binds to HDAC6, leading to reduced HDAC6 levels. However, overexpression of HDAC6 nullified IPA's beneficial effects, indicating the involvement of HDAC6/NOX2 signaling in IPA's action (63). These findings highlight that IPA can mitigate oxidative stress, inflammation and apoptosis in cardiomyocytes by inhibiting HDAC6/NOX2 signaling, demonstrating the potential therapeutic benefits of gut microbiota metabolites like IPA in heart failure treatment (Fig. 2).
Uric acid (UA) accumulation can lead to endothelial dysfunction, oxidative stress and inflammation (64,65). HDACs play a vital role in controlling the progression of pathological conditions in various illnesses. In a study, human umbilical vein endothelial cells were exposed to TSA or had HDAC6 levels reduced, resulting in a reduction of vascular endothelial cell injury caused by UA and an increase in fibroblast growth factor (FGF)21 expression, as well as the activation of AKT, endothelial nitric oxide synthase (eNOS) and forkhead box (Fox)O3a. The protective effects were negated by FGF21 knockdown. In live animals, both TSA and TubA decreased inflammation and tissue damage while enhancing FGF21 production and the activation of AKT, eNOS and FoxO3a in the aortic and kidney tissues of mice with high levels of UA (66). These findings indicate that HDACs, particularly HDAC6 inhibitors, mitigate UA-induced endothelial dysfunction by upregulating FGF21, which in turn activates the PI3K/AKT pathway. Therefore, focusing on HDAC6 shows promise as a treatment approach for UA-induced endothelial dysfunction.
The ISO-induced mouse heart-failure model reproduces sympathetic over-drive but lacks the metabolic and haemodynamic complexity of human heart failure with preserved ejection fraction. Similarly, telomere-shortening studies rely on young mice with short lifespans and rodent-specific telomerase kinetics, limiting translational value. Larger, multi-ethnic cohorts with pharmacokinetic/pharmacodynamic (PK/PD) sampling and off-target cardiac safety monitoring are still lacking.
HDAC6 and retinopathy
The social, health and economic burdens caused by retinal or macular degeneration-induced blindness are significant (67). New therapeutic targets and interventions are urgently needed for the more common atrophic ('dry') age-related macular degeneration (AMD), despite the availability of treatments for neovascular ('wet') AMD (68). Similarly, most inherited retinal diseases lack effective treatments. Although macular and retinal degeneration have genetic and clinical distinctions, they both exhibit similar pathological characteristics, including photoreceptor degeneration, retinal pigment epithelial atrophy, oxidative stress, hypoxia and autophagy deficiencies (69). A study discovered that zebrafish lacking atp6v0e1 and treated with tertiary statin A, an HDAC6 inhibitor, experienced notable enhancements in both photoreceptor outer segment size and visual acuity (70). Furthermore, retinal samples from rd10/rd10 mice that received tertiary statin A showed a significant increase in the quantity of cone photoreceptors located in the outer segments. In vitro studies indicated that ATP6V0E1 influenced hypoxia-inducible factor (HIF)-1α activity, although HDAC6 inhibitors did not significantly impact HIF-1α in the retina (70). Proteomic analysis revealed that the vision restoration mediated by HDAC6 inhibitors was associated with altered expression in ubiquitin-proteasome, phototransduction, metabolism and phagosomal pathways.
Researchers found increased HDAC6 levels and activity in the retinas of human diabetic postmortem donors, streptozotocin (STZ) rats and HuREC cells exposed to high glucose levels in a study using STZ rats as a model for type 1 diabetic retinopathy. Administering TubA, a specific HDAC6 inhibitor, successfully halted the rise in retinal microvascular hyperpermeability and inflammatory markers (71). Additionally, TubA treatment in STZ-induced diabetic rats reduced aging markers, restored the levels and function of sirtuin 1 and decreased the concentrations of ROS and oxidative stress markers such as 4-hydroxynonenal and nitrotyrosine. TubA's inhibition of HDAC6 was linked to the preservation of nuclear factor erythroid 2-related factor 2 (Nrf2)-dependent gene expression and the enhancement of thioredoxin-1 activity, ultimately strengthening its antioxidant properties. The antioxidant benefits of TubA inhibiting HDAC6 in diabetic retinas were confirmed by in vitro research conducted on HuREC cells exposed to glucose stress, which aligned with the in vivo outcomes (71). Therefore, the activation of HDAC6 is crucial in the development of oxidative and nitrosative stress caused by high blood sugar levels in the eye, leading to damage in small blood vessels and possibly diabetic retinopathy.
The impact of HDAC6 upregulation on ARPE-19 cells was investigated by researchers in both normal glucose and high glucose (HG) environments. In HG-stimulated cells, it was discovered that CAY10603 (Cay) treatment led to a significant reduction in intracellular levels of ROS (72,73). The therapy resulted in lower levels of malondialdehyde and myeloperoxidase, as well as increased activity of antioxidant enzymes like superoxide dismutase and catalase. Furthermore, Cay reduced the concentrations of inflammatory cytokines such as TNF-α, IL-1β, IL-6 and monocyte chemoattractant protein-1 in the cellular fluid (72). The therapy notably decreased apoptosis in ARPE-19 cells, as demonstrated by elevated Bcl-2 levels and reduced cleaved caspase-3 and cleaved caspase-9 levels. In addition, Cay decreased the levels of phospho-NF-κB p65, phospho-inhibitor of NF-κBα, NLRP3, cleaved caspase-1 and apoptosis-associated speck-like protein containing a CARD, while simultaneously raising the levels of cytoplasmic NF-κB p65 (72). The findings indicate that Cay may have potential as a treatment for diabetic retinopathy by reducing oxidative stress, inflammation and apoptosis in ARPE-19 cells through its effects on the NF-κB and NLRP3 inflammasome pathways induced by HG.
Although promising results have been reported using STZ-induced diabetic rats and ARPE-19 cell lines, these models only partially replicate the microvascular and neuro- inflammatory complexities of human diabetic retinopathy. Furthermore, the glucose-induced stress in vitro may not fully mimic chronic diabetic conditions in vivo. In addition, interspecies differences in retinal structure and immune response may limit the applicability of these findings to human patients. Future studies using retinal organoids or patient-derived cells may provide more clinically relevant insights.
Function of HDAC6 in liver injury
Depletion of glutathione (GSH), mitochondrial damage and oxidative stress are key factors in the development of acetaminophen (APAP) hepatotoxicity (74-76). A study has indicated that APAP-induced liver tissues and AML-12 cells show significantly increased HDAC6 expression and decreased malate dehydrogenase 1 (MDH1) levels. The novel HDAC6 inhibitor LT-630 has shown potential as a treatment for APAP-induced liver injury. LT-630 was found to enhance both the expression and acetylation of MDH1. When MDH1 was overexpressed in APAP-stressed AML-12 cells, there was an increase in the NADPH/NADP+ ratio, GSH levels were elevated and apoptosis was reduced (77). Notably, the beneficial effects of LT-630 were reversed when MDH1 was silenced with small inhibitory RNA (77). Thus, LT-630 appears to protect against liver damage by modulating MDH1 and reducing oxidative stress caused by APAP. The hepatoprotective effects of LT-630 and HDAC6 inhibition in acetaminophen-induced liver injury were evaluated primarily in murine hepatocytes and acute liver damage models. However, APAP hepatotoxicity in humans can exhibit substantial interindividual variability due to genetic, metabolic and environmental factors. Furthermore, acute liver failure models may not predict responses in chronic liver disease settings. More diverse and chronic liver models are needed, along with PK profiling of HDAC6 inhibitors in hepatic tissue.
Function of HDAC6 in sepsis
Sepsis, a serious illness caused by the body's overwhelming response to infection, is a leading factor in the high rates of illness and death in intensive care units (78). This syndrome can lead to the failure of various organs, such as the lungs, kidneys and liver, potentially culminating in multiple organ dysfunction syndrome (79,80). Researchers discovered in an experiment utilizing a cecal ligation and puncture (CLP) model to induce sepsis that HDAC6 plays a role in the advancement of sepsis by reducing the levels of prohibitin 1 (PHB1). Blocking HDAC6 activity significantly mitigated the effects of CLP-induced sepsis by preventing mitochondrial dysfunction and lowering oxidant production, which in turn protected rats from oxidative damage. The downregulation of PHB1 by HDAC6 interfered with the mitochondrial respiratory chain, resulting in increased oxidant production and oxidative stress, leading to severe oxidative damage in multiple organs (81). Although the CLP-induced sepsis model is a widely used and clinically relevant murine model, it still differs from human sepsis in terms of immune system complexity, timing of interventions and disease heterogeneity. The role of HDAC6 in human sepsis remains poorly characterized and differences in mitochondrial responses among species may influence outcomes. To improve translational relevance, studies using human immune cells or sepsis patient samples are warranted.
Function of HDAC6 in lung ischaemia-reperfusion injury (LIRI)
LIRI occurs due to hypoxia followed by blood flow restoration in the lungs (82,83). Research indicates that LIRI increases both the activity and expression of HDAC6. Inhibiting HDAC6 has been shown to provide protection against various types of IRIs and offers protective effects in different lung injury models. Research on MLE-12 cells showed that blocking HDAC6 decreased apoptosis, oxidative stress, inflammation and mitochondrial dysfunction caused by hypoxia/reoxygenation (H/R) (84). The protective impact was facilitated by the stimulation of the Nrf2/heme oxygenase (HO-1) signaling pathway and the deactivation of the ERK/NF-κB signaling pathway. However, these protective effects were partially reversed when MLE-12 cells were pretreated with the Nrf2 inhibitor ML385 or the ERK activator LM22B-10. Therefore, blocking HDAC6 can reduce lung epithelial cell damage caused by H/R by activating the Nrf2/HO-1 pathway and deactivating the ERK/NF-κB pathway (85). The findings from H/R cell models and murine LIRI studies demonstrate the potential of HDAC6 inhibition in mitigating oxidative lung injury. However, these models represent acute injury phases and may not reflect chronic lung injury, co-morbidities or the surgical and ventilatory complexities present in human transplantation contexts. Validation in larger animal models and ex vivo perfused human lungs could provide stronger translational evidence.
Function of HDAC6 in osteoarthritis
Osteoarthritis, a prevalent degenerative joint condition in older individuals, is characterized by cartilage damage, inflammation of the synovial membrane and hardening of the bone just beneath the cartilage (86,87). These pathological changes lead to chronic joint pain and dysfunction, significantly affecting quality of life and increasing healthcare costs (87). Researchers have found elevated levels of HDAC6 in the cartilage of osteoarthritic mice and in tert-butyl hydroperoxide (TBHP)-treated chondrocytes in vitro. Treatment with TubA, an HDAC6 inhibitor, effectively reduced HDAC6 expression, alleviated oxidative stress and lowered levels of apoptotic proteins, thereby promoting chondrocyte survival and preventing extracellular matrix degradation (88). Additionally, TubA-induced HDAC6 inhibition activated autophagy in chondrocytes, while autophagy inhibitors negated TubA's protective effects (88). The results suggest that targeting HDAC6 could be a potential treatment option for osteoarthritis. However, while HDAC6 inhibition reduced oxidative damage and preserved cartilage in mouse models and TBHP-treated chondrocytes, the spontaneous and slowly progressive nature of human osteoarthritis is not fully captured in these systems. Rodent joints (compared with human joints) differ in size, biomechanics and immune response (89,90). In addition, therapeutic timing and long-term outcomes remain underexplored. Studies using human chondrocytes or advanced 3D joint-on-chip models could help address these gaps.
Function of HDAC6 in cytotoxicity
Nanoplastics (NPs), originating from microplastics, present potential health hazards. Polystyrene (PS)-NPs of 100 nm can penetrate cells, including mouse embryonic fibroblasts (MEFs), where their accumulation leads to inflammation and oxidative stress (91,92). PS-NPs are internalized via endocytosis and predominantly gather at juxtamembranous sites, avoiding the nucleus, and are eventually cleared from cells after exposure stops (93,94). Their proximity to the nucleus is likely due to retrograde transport along microtubes. Researchers discovered that treating PS-NP-exposed MEFs with inhibitors of HDAC6, dynamin or microtubule polymerization markedly reduced both intracellular and intranuclear accumulation (95). Furthermore, HDAC6 inhibitors facilitated the rapid clearance of PS-NPs. Blocking retrograde transport also diminished the activation of antioxidant response pathways, inflammation, oxidative stress and ROS production (95). Therefore, inhibiting the reverse transport of non-biodegradable PS-NPs promotes their quick export via exocytosis, thereby reducing cytotoxicity. The study of PS-NPs in murine embryonic fibroblasts provides a useful model for nanotoxicity, but their intracellular behavior, clearance kinetics and interactions with the immune system in humans are still poorly understood. Furthermore, in vitro systems lack the systemic complexity and exposure routes of environmental NP exposure. Bridging in vitro findings with in vivo toxicology and human exposure assessment will be crucial for real-world risk evaluation.
HDAC6 and cancer
Role of HDAC6 in cancer by influencing oxidative stress
HDAC6 is gaining recognition for its significant role in cancer, particularly through its impact on oxidative stress. HDAC6 regulates the acetylation of various non-histone proteins, affecting cellular redox balance, protein degradation and stress responses. In cancer, HDAC6 is often upregulated, increasing oxidative stress, which in turn promotes tumor progression and resistance to therapy. The enzyme's regulation of ROS production, mitochondrial function and autophagy highlights its critical role in cancer biology. Targeting HDAC6 with specific inhibitors has demonstrated potential in reducing tumor growth and overcoming chemoresistance by mitigating oxidative stress and enhancing cellular homeostasis. This section reviews HDAC6's role in cancer development and progression through its influence on oxidative stress, emphasizing the therapeutic potential of HDAC6 inhibition in oncology.
Oral squamous cell carcinoma (OSCC)
Resistance to chemotherapy in OSCC presents a major obstacle, frequently leading to cancer recurrence and metastasis (96,97). Cancer stem cell (CSC) subpopulations are notably resistant to treatment due to epigenetic regulation mechanisms (98). HDACs are crucial epigenetic regulators of gene expression, with HDAC6 playing a role in processes such as oxidative stress, autophagy and the DNA damage response (99). Researchers found that HDAC6 accumulates in cisplatin-resistant (CisR) cell lines and CSCs, which showed reduced DNA damage and ROS levels, along with increased expression of PRDX2. Treatment with TubA, a specific HDAC6 inhibitor, heightened oxidative stress and DNA damage while reducing PRDX2 levels (100). Additionally, TubA induced apoptosis and diminished the stemness phenotype in both CisR and CSC cells (100). The results indicate that targeting HDAC6 with drugs such as TubA might be able to address chemoresistance and decrease CSC characteristics in OSCC.
Lung cancer
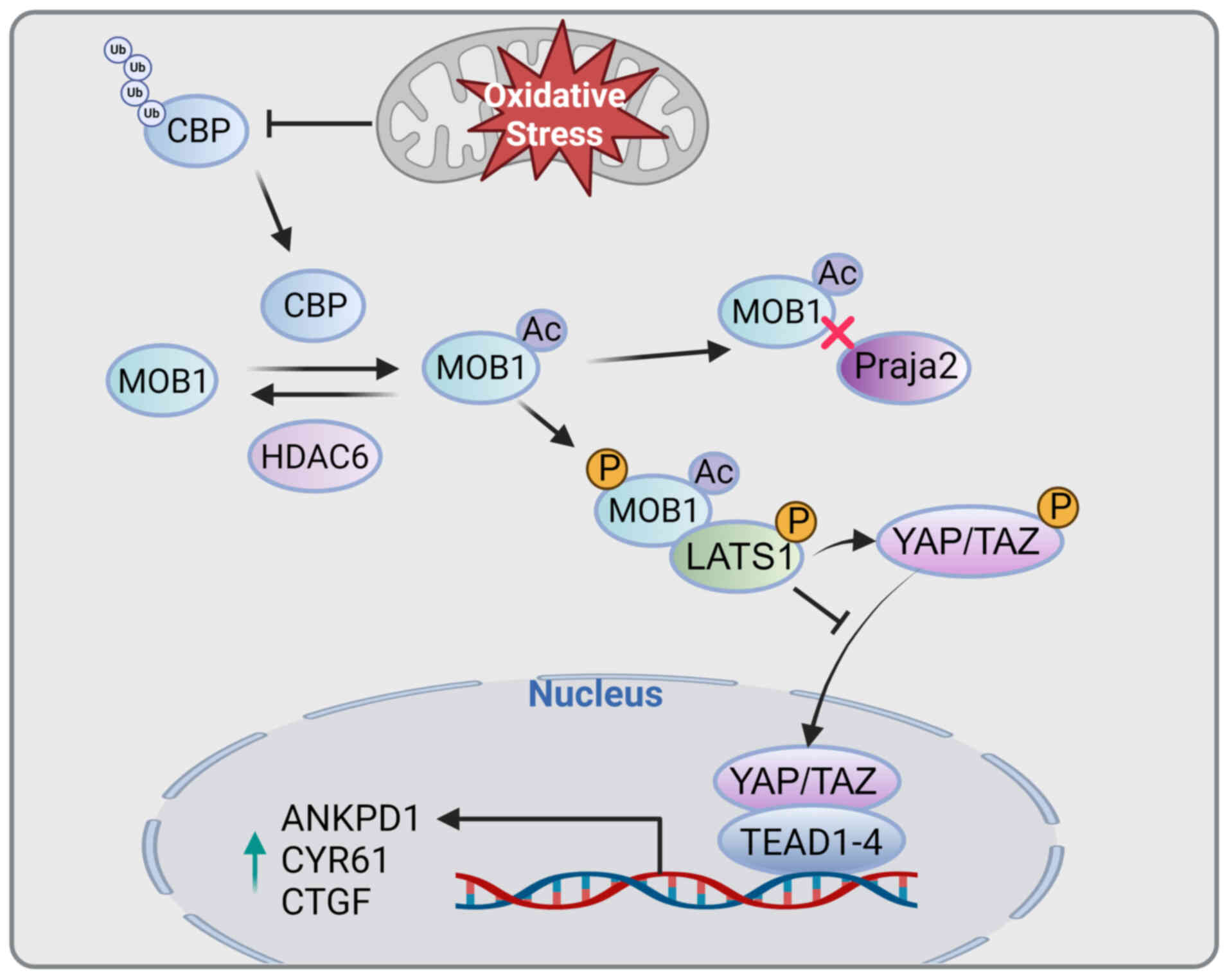
Cells constantly produce ROS, which can lead to oxidative stress if their concentrations become excessive (101). ROS play a crucial role in regulating the Hippo pathway (102). Mps one binder 1 (MOB1), an essential component of the Hippo signaling pathway, is phosphorylated by mammalian STE20-like protein kinase 1/2 (MST1/2) and enhances the activity of large tumor suppressor kinase 1/2 (LATS1/2) (103). It interacts with the acetyltransferase CREB-binding protein (CBP) and gets acetylated at lysine 11 (MOB1-K11) (104). Conversely, HDAC6 deacetylates MOB1. When MOB1 is acetylated at K11, its binding affinity for the E3 ligase Praja2 decreases, leading to its ubiquitination and stabilization. The acetylation of MOB1 increases its phosphorylation, leading to the activation of LATS1. Oxidative stress inhibits the degradation of CBP, leading to the acetylation of MOB1, which is consistent with the activity of MST1/2 kinases and opposes the deacetylation effect of HDAC6. This links oxidative stress to Hippo pathway activation. In vitro, the mutant MOB1-K11R, which lacks acetylation, enhances the proliferation, migration and invasion of lung cancer cells, and in vivo, it accelerates tumor growth compared to wild-type MOB1 (105). The interaction between oxidative stress and CBP controls the acetylation of MOB1-K11, leading to the activation of LATS1 and the Hippo pathway. This activation prevents the nuclear translocation of yes-associated protein/transcriptional co-activator with PDZ-binding motif and hinders tumor progression (Fig. 3).

In non-small cell lung cancer (NSCLC) with KRAS mutations, changes in liver kinase B1 (LKB1) frequently result in a worse prognosis than mutations in TP53 (106). LKB1 plays a vital role as a tumor suppressor, regulating multiple signaling pathways in response to energy deprivation (106). A recent study on blocking HDAC6 using both pharmacological and genetic techniques revealed its impact on the function of various key glycolytic enzymes. Researchers used mouse cell lines with KRAS/LKB1 (KL) and KRAS/TP53 mutations to account for the variability caused by germline and somatic mutations in human models. They examined the metabolic characteristics and responses to HDAC6 blockade, assessing the impact on cancer cell proliferation in vitro and tumor development in vivo. The findings suggested that KL mutant cells possessed intrinsically reduced levels of redox-responsive coenzymes, rendering them more susceptible to the HDAC6 inhibitor ACY-1215 (107). This vulnerability was due to their diminished capacity to ramp up compensatory metabolic processes and manage oxidative stress. Furthermore, the concurrent use of HDAC6 blockers and glutaminase inhibitors resulted in a notable increase in cell mortality and enhanced the body's ability to combat tumors in a KL lung cancer models, both in vitro and in vivo (107). Thus, targeting HDAC6 presents a potential opportunity in KL NSCLC by exploiting the cells' weakened capacity to handle glycolytic dysfunction, offering a promising strategy for managing KRAS-mutant NSCLC with concurrent LKB1 mutations.
Glioblastoma (GBM)
GBM is a challenging brain tumor that is both aggressive and drug-resistant, characterized by genetically unstable and invasive cells that pose significant treatment difficulties (108,109). This has driven the need for new therapeutic approaches. A group of scientists have studied a range of hydroxamic acids containing abiraterone as possible dual blockers of cytochrome P450 family 17 subfamily A member 1 (CYP17A1) and HDAC6 to treat GBM. Among these, Compound 12 stood out for its ability to induce apoptosis, suppress genes linked to tumor recurrence, increase oxidative stress and trigger DNA damage responses (110). Molecular modeling studies confirmed that Compound 12 has strong inhibitory effects on both CYP17A1 and HDAC6. Compound 12 was found to efficiently decrease tumor growth in vivo in xenograft and orthotopic mouse models, with no notable adverse effects (110). These findings highlight the potential of dual CYP17A1 and HDAC6 inhibition as a promising strategy to overcome resistance in GBM therapy, offering a new avenue for more effective treatments.
Role of HDAC6 in other cancers
The multifunctional roles of HDAC6 extend far beyond its involvement in neurodegenerative diseases, cardiovascular disorders and metabolic conditions, as its influence is significantly noted across various types of cancers. Previously, it has been shown that HDAC6 has a role in the development and advancement of liver (111), breast (112), colorectal (113,114), gastric (115), ovarian and endometrial cancer (116). These findings emphasized the importance of HDAC6 in regulating responses to oxidative stress, cytoskeletal dynamics, epithelial-mesenchymal transition, protein homeostasis and intercellular communication, all of which play crucial roles in cancer progression and metastasis (117-120). HDAC6 can remove acetyl groups from non-histone proteins like α-tubulin (121), HSP90 (122), cortactin (123) and various transcription factors, enabling it to control essential mechanisms that cancer cells use to survive, grow and spread. The inhibition of HDAC6 has shown promising therapeutic potential in preclinical models by disrupting these oncogenic processes, offering new avenues for cancer treatment (124,125). Building on this foundation, it is crucial to explore the roles of HDAC6 in other cancer types, further elucidating its broad impact on tumorigenesis and its potential as a therapeutic target. HDAC6's influence spans across a diverse array of malignancies, including bladder, pancreatic (126), cervical and gallbladder cancer (127). In these cancers, HDAC6 modulates oxidative stress, enhances cell motility and affects key signaling pathways that are pivotal for cancer cell survival and resistance to therapy (127-130). The enzyme's regulatory functions often intersect with vital cellular mechanisms such as autophagy, apoptosis and immune response modulation, underscoring its central role in maintaining the malignant phenotype (Table I).
HDAC6 is a potential therapeutic target for disease
Due to its diverse functions in controlling oxidative stress and preserving cellular balance, HDAC6 has become a potential target for treating various conditions marked by redox disruption, protein misfolding and inflammation. The development of specific HDAC6 inhibitors offers new opportunities for treating conditions such as neurodegenerative diseases, cardiovascular disorders, metabolic conditions and cancer (131-135) (Table II). This chapter discusses some of the therapeutic strategies and examples of HDAC6 inhibitors that have shown promise in preclinical and clinical studies (Table III).
TubA
A potent HDAC6 inhibitor, TubA, has shown protective effects against multiple oxidative stress-related diseases (136). As an example, HDAC6 can be found constitutively in the mouse retina as well as in the cone-like mouse cell line 661W. HDAC6 can be inhibited by the specific inhibitor TubA, leading to acetylation of α-tubulin, HDAC6's primary substrate (137). When hydrogen peroxide produces oxidative stress, TubA promotes cell survival and activates heat shock transcription factor 1, activating HSP70 and HSP25 (137). In addition, oxidative stress inhibits the peroxiredoxin 1 (Prx1) redox-regulated protein in 661W cells. Prx1's peroxide-reducing activity depends on its acetylation, which HDAC6 inhibits (37). Photoreceptors may be protected from damage by Prx1-retained activity after preincubation with TubA. An analysis of hereditary vision loss in zebrafish (dyeucd6) was carried out to determine whether TubA treatment has a therapeutic effect on visual function. The application of TubA in vivo led to hyperacetylation of alpha-microtubulin, as well as the ability to rescue visual function and retinal morphology (70,137). Therefore, inhibiting HDAC6 and modulating peroxiredoxin activity may play an important role in protecting retinal cells, particularly photoreceptors, which are exposed to high levels of ROS.
Tubacin
Tubacin is a selective HDAC6 inhibitor that has demonstrated protective effects in models of cardiovascular disease. Tubacin treatment in endothelial cells decreased ROS generation by blocking NADPH oxidase function and boosting eNOS activity, resulting in elevated NO levels (138). This led to improved endothelial function and reduced vascular inflammation. Tubacin's ability to stabilize microtubules by inhibiting HDAC6-mediated deacetylation of α-tubulin also contributes to its protective effects on the endothelium, promoting better cellular structure and function. In atherosclerosis models, Tubacin has been shown to reduce plaque formation and stabilize existing plaques by decreasing oxidative stress and inflammation (139). The compound's capacity to regulate the NF-κB and MAPK signaling pathways, crucial in inflammatory reactions, enhances its promise as a treatment for cardiovascular conditions.
Ricolinostat
It has been determined that ACY-1215 (Ricolinostat) provides the most effective treatment for macrophages activated by LPS (41,140,141). The treatment dose was determined using flow cytometry. In a study using RAW264.7 macrophages, controls, LPS-treated macrophages and ACY-1215-treated macrophages were compared. LPS-treated cells were treated with ACY-1215 (10 µM) 2 h before LPS treatment, and ROS, inflammatory cytokines, mitochondrial ultrastructure, mitochondrial membrane potential, RNA and protein expression were determined after 24 h of LPS treatment. In addition, HDAC6 knockdown was investigated in relation to the inflammatory response of LPS-activated RAW264.7 macrophages. It was demonstrated that inhibition of HDAC6 suppressed the production of ROS and inhibited the expression of pro-inflammatory cytokines in LPS-activated RAW264.7 cells, including TNF-α, IL-1 and IL-6, in the cells (41). Furthermore, ACY-1215 restored the mitochondrial membrane potential and ultrastructure of LPS-activated macrophages to normal levels (41). As well as inhibiting HDAC6 expression, ACY-1215 activated MAPK and NF-κB signaling pathways by normalizing Toll-like receptor 4, Nrf2 and HO-1 protein expression (142).
Vorinostat
Vorinostat, also called SAHA, is a broad-spectrum HDAC inhibitor that has been authorized for treating cutaneous T-cell lymphoma (143). Although not selective for HDAC6, Vorinostat has shown potential to modulate oxidative stress and improve the efficacy of anticancer therapies. During preclinical research, Vorinostat therapy increased levels of ROS in malignant cells, resulting in increased cell death and decreased tumor cell proliferation (144). The potential of Vorinostat as a therapeutic agent for various cancers is supported by its ability to inhibit multiple HDACs and regulate different cellular pathways related to oxidative stress and protein homeostasis (145). The compound's broad-spectrum activity makes it a valuable addition to the arsenal of anticancer therapies, particularly in cases where selective HDAC6 inhibitors may not be sufficient.
PK and toxicity of HDAC6 inhibitors
HDAC6 inhibitors such as TubA, ACY-1215 (ricolinostat) and tubacin have shown therapeutic efficacy in preclinical models of oxidative stress-related diseases. However, a thorough understanding of their PK and toxicity is critical for clinical translation.
TubA exhibits high selectivity for HDAC6 with nanomolar potency. In murine models, it shows moderate bioavailability when administered intraperitoneally and a relatively short half-life (~1-2 h) due to rapid hepatic metabolism (146). Limited oral bioavailability has been reported, necessitating formulation optimization for systemic administration. Toxicological studies in rodents indicate that TubA is generally well tolerated at therapeutic doses, with minimal off-target effects (147). However, long-term toxicity and potential immunosuppressive effects require further investigation (147,148).
ACY-1215 (ricolinostat) is a clinically advanced HDAC6-selective inhibitor with favorable PK. Phase I clinical trials in patients with multiple myeloma and lymphoma revealed an elimination half-life of 3-5 h and good oral bioavailability (149,150). Ricolinostat was generally well tolerated, with the most common adverse effects being fatigue, diarrhea and transient cytopenias. These findings support its use in combination therapies. However, reversible inhibition of immune function and hepatic enzyme elevation have been observed in certain patients, underscoring the need for careful monitoring (151).
Tubacin, while effective in vitro, has drawbacks of poor metabolic stability and low systemic exposure in vivo, which limits its development as a clinical candidate. Its use has primarily been restricted to proof-of-concept studies due to its unfavorable PK profile (152,153).
Despite promising early-phase safety data, most HDAC6 inhibitors have not yet undergone large-scale toxicity studies in humans. The long-term effects on normal tissue homeostasis, off-target deacetylation events and potential reproductive toxicity remain largely unexplored. Further PK modeling and toxicity profiling in diverse species are essential to advance these compounds toward regulatory approval (153).
Challenges and limitations in HDAC6 inhibitor research
While HDAC6 inhibitors have shown promising therapeutic potential in preclinical models and early-stage clinical trials, several limitations exist that hinder their full clinical translation. These limitations encompass challenges in modeling human disease, drug development, PK and toxicity. Addressing these gaps will be crucial for the successful application of HDAC6 inhibitors in clinical practice.
Preclinical models
Although numerous animal models, including rodent models of neurodegeneration, cancer and cardiovascular diseases, have been used to assess the efficacy of HDAC6 inhibitors, these models often fail to fully recapitulate the complexity of human diseases (154,155). For instance, rodent models typically do not mimic the slow, multifactorial progression of diseases like Parkinson's or Alzheimer's (139). Similarly, the short lifespan and lack of sex- and age-related variability in animal models limit their ability to predict long-term therapeutic outcomes in humans (156). Furthermore, these models often do not account for the impact of comorbidities and multifactorial conditions that are common in human patients, complicating the translation of preclinical findings to human treatments (156).
Lack of long-term toxicity data
Most current research on HDAC6 inhibitors is based on short-term studies and long-term toxicity data remain scarce (157). Chronic inhibition of HDAC6, particularly in neuronal cells, may impair autophagic processes and protein degradation, leading to potential neurotoxicity (158). The long-term effects of HDAC6 inhibition on other organ systems, such as the heart, liver and kidneys, are largely unknown. Studies investigating off-target effects and cumulative toxicity are essential for assessing the safety of these compounds in prolonged use (137).
PK/PD variability
The PK profiles of HDAC6 inhibitors, particularly in humans, are still not fully understood. Variability in oral bioavailability, half-life and hepatic metabolism can affect both the efficacy and safety of these drugs. For instance, certain inhibitors, such as TubA, show promising preclinical efficacy but have been associated with high systemic exposure and off-target effects at therapeutic doses, raising concerns about their safe application in humans. Furthermore, drug-drug interactions remain a significant issue, particularly in the context of patients taking multiple medications for complex conditions like cancer or cardiovascular disease (159).
Limited clinical data
Clinical trials (NCT01997840, NCT02189343, NCT02091063, NCT03176472) of HDAC6 inhibitors, such as Ricolinostat (ACY-1215), have shown encouraging results in terms of target engagement and initial safety, but large-scale, randomized controlled trials are still lacking (137,149). The absence of longitudinal studies means that the efficacy of these inhibitors in real-world clinical settings remains uncertain. Furthermore, phase II and III trials focusing on hard clinical endpoints (e.g., survival, disease progression) are essential to confirm the long-term benefits of HDAC6 inhibitors in treating neurodegenerative diseases, cancer and cardiovascular disorders.
Challenges in drug development and biomarker identification
A significant challenge in HDAC6 inhibitor development is the lack of reliable biomarkers that can measure drug efficacy and predict patient response. For instance, biomarkers like acetyl-α-tubulin have been suggested, but their use in clinical trials is still under investigation. The absence of predictive biomarkers complicates patient stratification and limits the ability to tailor treatment to specific genetic profiles or disease stages (153,160,161).
Safety concerns in vulnerable populations
Another limitation is the lack of data on the safety of HDAC6 inhibitors in vulnerable populations, including elderly patients, children and those with comorbidities. The effect of HDAC6 inhibition on reproductive health, immune function and neurological development is still under-researched, and these concerns will need to be addressed through dedicated clinical trials.
Conclusion and future perspectives
HDAC6 is a pivotal regulator of oxidative stress and cellular homeostasis. By modulating mitochondrial dynamics, antioxidant defences, protein-quality control, stress responses and multiple signalling cascades, it emerges as a compelling therapeutic target for diseases driven by redox imbalance, protein misfolding and inflammation (Fig. 4). Selective HDAC6 inhibitors, such as TubA, ACY-1215 (ricolinostat), Tubacin and ACY-738, have already shown pre-clinical efficacy across neurodegenerative, cardiovascular, metabolic and oncological models by reducing oxidative damage, restoring proteostasis and improving cell survival (162). Combination treatments that focus on various pathways may provide improved therapeutic advantages due to the intricate characteristics of conditions like neurodegeneration, cancer and metabolic disorders. Combining HDAC6 inhibitors with other treatments, such as proteasome inhibitors, immunotherapies or conventional chemotherapies, may enhance efficacy and overcome resistance. Preclinical studies and clinical trials exploring these combinations could provide valuable insight into synergistic effects and optimal therapeutic strategies. Identifying biomarkers that predict response to HDAC6 inhibition could improve patient stratification and personalized medicine approaches. Biomarkers such as unique protein acetylation profiles, levels of ROS or genetic mutations linked to HDAC6 function could help identify patients who may benefit from HDAC6-targeted treatments. Incorporating biomarker studies into clinical trials may improve the effectiveness and safety of these therapies. Beyond the current focus on neurodegenerative diseases, cardiovascular diseases, metabolic disorders and cancer, there is potential to explore HDAC6 inhibitors in other conditions characterized by oxidative stress and protein misfolding. For instance, inflammatory diseases, autoimmune disorders and infectious diseases could benefit from therapies targeting HDAC6. Investigating these new therapeutic avenues could broaden the impact of HDAC6 inhibitors in medicine.
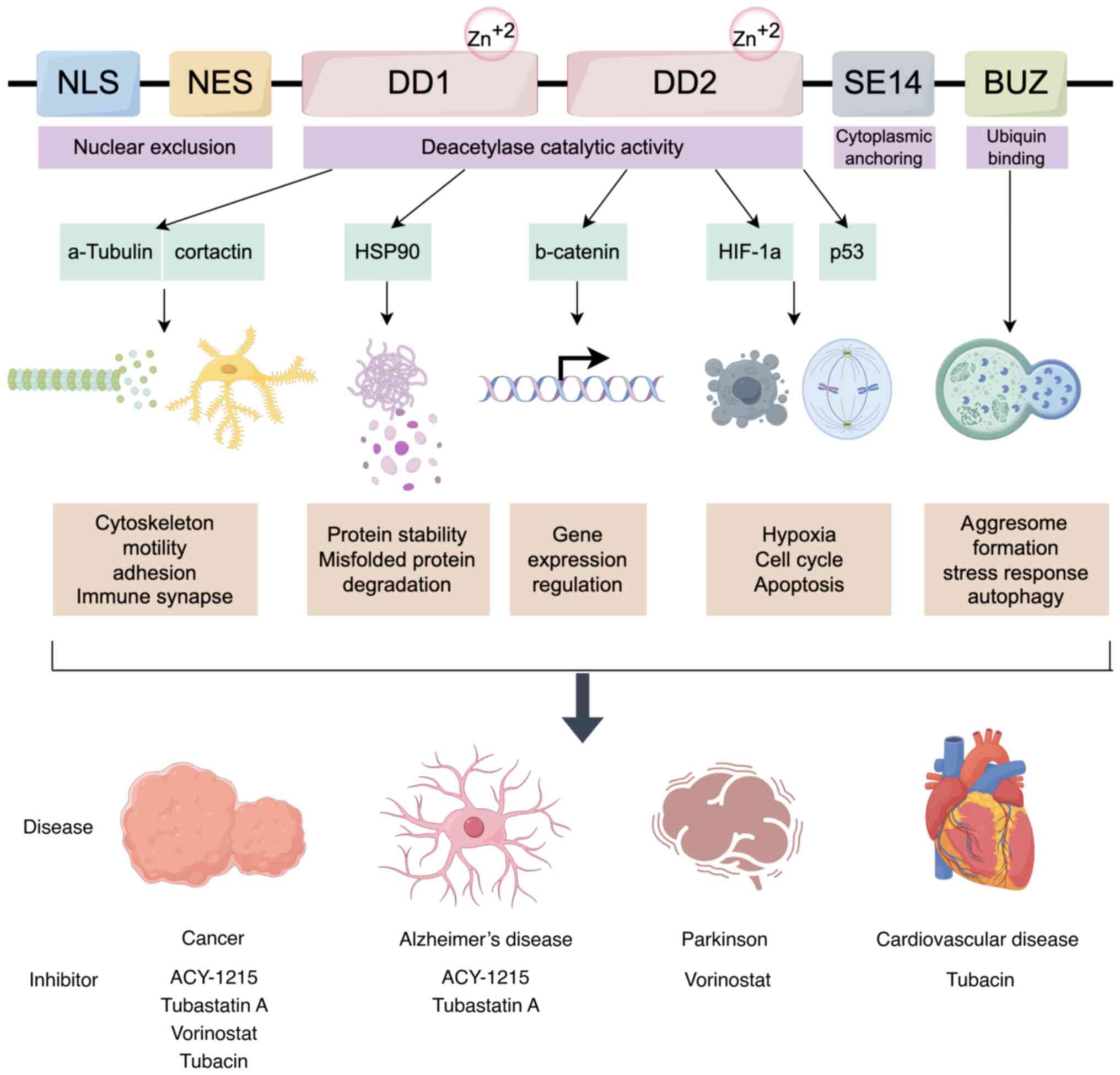
HDAC6 stands out as a critical enzyme involved in maintaining cellular homeostasis, particularly under oxidative stress conditions. Its unique structural and functional properties make it a promising target for therapeutic intervention across a range of diseases. The development and optimization of selective HDAC6 inhibitors offer significant potential for improving treatment outcomes in neurodegenerative diseases, cardiovascular disorders, metabolic diseases, and cancer. Looking ahead, continued research into HDAC6's mechanisms of action, the development of highly selective inhibitors, exploration of combination therapies and identification of predictive biomarkers will be crucial for translating preclinical success into clinical practice. New scientific advancements can help overcome these obstacles, leading to HDAC6-targeted treatments that may greatly improve patient outcomes and quality of life for those suffering from these challenging and disabling conditions.
Despite promising preclinical findings, current HDAC6-targeted strategies face several limitations. First, most studies are based on in vitro models or rodent experiments, which may not fully replicate human disease complexity, PK or immune responses. Second, clinical data-particularly in terms of long-term safety, toxicity and off-target effects-remain scarce. For instance, although HDAC6 inhibitors such as ACY-1215 (ricolinostat) have entered early-phase clinical trials (137,149), data on their use in oxidative stress-related disorders are limited. Furthermore, adverse effects, such as cytopenias and potential hepatic toxicity, have been reported and warrant further investigation. Future research should emphasize the development of organ-specific delivery strategies, long-term toxicity assessment and clinical biomarker validation to guide patient selection and therapeutic monitoring. Addressing these gaps will accelerate the translation of HDAC6-targeted strategies from bench to bedside, offering new options for patients with currently intractable oxidative stress-related diseases.
Availability of data and materials
Not applicable.
Authors' contributions
FQ conducted the literature search, organized the information and participated in the writing of the manuscript. QZ participated in the drawing of the article's figures as well as in manuscript preparation. YJ wrote the manuscript and provided supervision. All authors have read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 82401009) and the Research Start-up Fee for Introducing Talents to Jinan Central Hospital (grant no. YJRC2023001).
References
|
Forman HJ and Zhang H: Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat Rev Drug Discov. 20:689–709. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Teleanu DM, Niculescu AG, Lungu II, Radu CI, Vladâcenco O, Roza E, Costăchescu B, Grumezescu AM and Teleanu RI: An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int J Mol Sci. 23:59382022. View Article : Google Scholar : PubMed/NCBI | |
|
Jiang H, Zuo J, Li B, Chen R, Luo K, Xiang X, Lu S, Huang C, Liu L, Tang J and Gao F: Drug-induced oxidative stress in cancer treatments: Angel or devil? Redox Biol. 63:1027542023. View Article : Google Scholar : PubMed/NCBI | |
|
Wang X, Zhang G, Dasgupta S, Niewold EL, Li C, Li Q, Luo X, Tan L, Ferdous A, Lorenzi PL, et al: ATF4 protects the heart from failure by antagonizing oxidative stress. Circ Res. 131:91–105. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Masenga SK, Kabwe LS, Chakulya M and Kirabo A: Mechanisms of oxidative stress in metabolic syndrome. Int J Mol Sci. 24:78982023. View Article : Google Scholar : PubMed/NCBI | |
|
Shi X, Zhou H, Wei J, Mo W, Li Q and Lv X: The signaling pathways and therapeutic potential of itaconate to alleviate inflammation and oxidative stress in inflammatory diseases. Redox Biol. 58:1025532022. View Article : Google Scholar : PubMed/NCBI | |
|
Guan D, Men Y, Bartlett A, Hernández MAS, Xu J, Yi X, Li HS, Kong D, Mazitschek R and Ozcan U: Central inhibition of HDAC6 re-sensitizes leptin signaling during obesity to induce profound weight loss. Cell Metab. 36:857–876.e10. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Kim JY, Hwang HG, Lee JY, Kim M and Kim JY: Cortactin deacetylation by HDAC6 and SIRT2 regulates neuronal migration and dendrite morphogenesis during cerebral cortex development. Mol Brain. 13:1052020. View Article : Google Scholar : PubMed/NCBI | |
|
Wu TY, Chen M, Chen IC, Chen YJ, Chen CY, Wang CH, Cheng JJ, Nepali K, Chuang KH and Liou JP: Rational design of synthetically tractable HDAC6/HSP90 dual inhibitors to destroy immune-suppressive tumor microenvironment. J Adv Res. 46:159–171. 2023. View Article : Google Scholar : | |
|
Zhang S, Sui L, Kong X, Huang R and Li Z: HDAC6 decreases H4K16 and α-tubulin acetylation during porcine oocyte maturation. Cell Cycle. 22:2057–2069. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Liang T, Qi C, Lai Y, Xie J, Wang H, Zhang L, Lin T, Jv M, Li J, Wang Y, et al: HDAC6-mediated α-tubulin deacetylation suppresses autophagy and enhances motility of podocytes in diabetic nephropathy. J Cell Mol Med. 24:11558–11572. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Luthold C, Varlet AA, Lambert H, Bordeleau F and Lavoie JN: Chaperone-assisted mitotic actin remodeling by BAG3 and HSPB8 involves the deacetylase HDAC6 and its substrate cortactin. Int J Mol Sci. 22:1422020. View Article : Google Scholar : PubMed/NCBI | |
|
Du Y, Yang X, Li Z, Le W, Hao Y, Song Y, Wang F and Guan Y: HDAC6-mediated Hsp90 deacetylation reduces aggregation and toxicity of the protein alpha-synuclein by regulating chaperone-mediated autophagy. Neurochem Int. 149:1051412021. View Article : Google Scholar : PubMed/NCBI | |
|
Bai P, Mondal P, Bagdasarian FA, Rani N, Liu Y, Gomm A, Tocci DR, Choi SH, Wey HY, Tanzi RE, et al: Development of a potential PET probe for HDAC6 imaging in Alzheimer's disease. Acta Pharm Sin B. 12:3891–3904. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao J, He Y, Duan Y, Ma Y, Dong H, Zhang X, Fang R, Zhang Y, Yu M and Huang F: HDAC6 deficiency has moderate effects on behaviors and Parkinson's disease pathology in mice. Int J Mol Sci. 24:99752023. View Article : Google Scholar : PubMed/NCBI | |
|
Ducellier S, Demeules M, Letribot B, Gaetani M, Michaudel C, Sokol H, Hamze A, Alami M, Nascimento M and Apcher S: Dual molecule targeting HDAC6 leads to intratumoral CD4+ cytotoxic lymphocytes recruitment through MHC-II upregulation on lung cancer cells. J Immunother Cancer. 12:e0075882024. View Article : Google Scholar : PubMed/NCBI | |
|
Ranjbarvaziri S, Zeng A, Wu I, Greer-Short A, Farshidfar F, Budan A, Xu E, Shenwai R, Kozubov M, Li C, et al: Targeting HDAC6 to treat heart failure with preserved ejection fraction in mice. Nat Commun. 15:13522024. View Article : Google Scholar : PubMed/NCBI | |
|
Wu YX, Li BQ, Yu XQ, Liu YL, Chui RH, Sun K, Geng DG and Ma LY: Histone deacetylase 6 as a novel promising target to treat cardiovascular disease. Cancer Innov. 3:e1142024. View Article : Google Scholar | |
|
Kundu S, Gairola S, Verma S, Mugale MN and Sahu BD: Chronic kidney disease activates the HDAC6-inflammatory axis in the heart and contributes to myocardial remodeling in mice: Inhibition of HDAC6 alleviates chronic kidney disease-induced myocardial remodeling. Basic Res Cardiol. 119:831–852. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Wu C, Pan Y, Wang L, Liu M, Tu P, Chen S, Shi L, Yan D, Ma Y and Guo Y: Inhibition of HDAC6 promotes microvascular endothelial cells to phagocytize myelin debris and reduces inflammatory response to accelerate the repair of spinal cord injury. CNS Neurosci Ther. 30:e144392024. View Article : Google Scholar : | |
|
Wen Y, Ye S, Li Z, Zhang X, Liu C, Wu Y, Zheng R, Xu C, Tian J, Shu L, et al: HDAC6 inhibitor ACY-1215 enhances STAT1 acetylation to block PD-L1 for colorectal cancer immunotherapy. Cancer Immunol Immunother. 73:72024. View Article : Google Scholar : PubMed/NCBI | |
|
Zhu Y, Feng M, Wang B, Zheng Y, Jiang D, Zhao L, Mamun MAA, Kang H, Nie H, Zhang X, et al: New insights into the non-enzymatic function of HDAC6. Biomed Pharmacother. 161:1144382023. View Article : Google Scholar : PubMed/NCBI | |
|
Kaur S, Rajoria P and Chopra M: HDAC6: A unique HDAC family member as a cancer target. Cell Oncol (Dordr). 45:779–829. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Yang J, Liu Y, Yin H, Xie S, Zhang L, Dong X, Ni H, Bu W, Ma H, Liu P, et al: HDAC6 deacetylates IDH1 to promote the homeostasis of hematopoietic stem and progenitor cells. EMBO Rep. 24:e560092023. View Article : Google Scholar | |
|
Peng J, Xie F, Qin P, Liu Y, Niu H, Sun J, Xue H, Zhao Q, Liu J and Wu J: Recent development of selective inhibitors targeting the HDAC6 as anti-cancer drugs: Structure, function and design. Bioorg Chem. 138:1066222023. View Article : Google Scholar : PubMed/NCBI | |
|
Izumi H, Kaneko Y and Nakagawara A: Molecular regulation of autophagy and asymmetric cell division by cancer stem cell marker CD133. Cells. 12:8192023. View Article : Google Scholar : PubMed/NCBI | |
|
Balmik AA, Sonawane SK and Chinnathambi S: The extracellular HDAC6 ZnF UBP domain modulates the actin network and post-translational modifications of Tau. Cell Commun Signal. 19:492021. View Article : Google Scholar : PubMed/NCBI | |
|
Calogero AM, Basellini MJ, Isilgan HB, Longhena F, Bellucci A, Mazzetti S, Rolando C, Pezzoli G and Cappelletti G: Acetylated α-tubulin and α-synuclein: physiological interplay and contribution to α-synuclein oligomerization. Int J Mol Sci. 24:122872023. View Article : Google Scholar | |
|
Ripa L, Sandmark J, Hughes G, Shamovsky I, Gunnarsson A, Johansson J, Llinas A, Collins M, Jung B, Novén A, et al: Selective and bioavailable HDAC6 2-(Difluoromethyl)-1,3,4-oxadiazole substrate inhibitors and modeling of their bioactivation mechanism. J Med Chem. 66:14188–14207. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Si L, Lai T, Zhao J, Jin Y, Qi M, Li M, Fu H, Shi X, Ma L and Guo R: Identification of a novel pyridine derivative with inhibitory activity against ovarian cancer progression in vivo and in vitro. Front Pharmacol. 13:10644852022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Z, Zhang X and Huang A: Aggresome-autophagy associated gene HDAC6 is a potential biomarker in pan-cancer, especially in colon adenocarcinoma. Front Oncol. 11:7185892021. View Article : Google Scholar : PubMed/NCBI | |
|
Lemos M and Stefanova N: Histone deacetylase 6 and the disease mechanisms of α-synucleinopathies. Front Synaptic Neurosci. 12:5864532020. View Article : Google Scholar | |
|
Dawood M, Hegazy MF, Elbadawi M, Fleischer E, Klinger A, Bringmann G, Kuntner C, Shan L and Efferth T: Vitamin K3 chloro derivative (VKT-2) inhibits HDAC6, activates autophagy and apoptosis, and inhibits aggresome formation in hepatocellular carcinoma cells. Biochem Pharmacol. 180:1141762020. View Article : Google Scholar | |
|
English K and Barton MC: HDAC6: A key link between mitochondria and development of peripheral neuropathy. Front Mol Neurosci. 14:6847142021. View Article : Google Scholar : PubMed/NCBI | |
|
Xia K, Qiu T, Jian Y, Liu H, Chen H, Liu X, Chen Z and Wang L: Degradation of histone deacetylase 6 alleviates ROS-mediated apoptosis in renal ischemia-reperfusion injury. Biomed Pharmacother. 165:1151282023. View Article : Google Scholar : PubMed/NCBI | |
|
Kim J, Jangili P, Kim J, Lucia SE, Ryu JR, Prasad R, Zi S, Kim P, Sun W and Kim JS: Mitochondrial NIR imaging probe mitigating oxidative damage by targeting HDAC6. Chem Commun (Camb). 59:10109–10112. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Xue Y, Gan B and Zhou Y, Wang T, Zhu T, Peng X, Zhang X and Zhou Y: Advances in the mechanistic study of the control of oxidative stress injury by modulating HDAC6 activity. Cell Biochem Biophys. 81:127–139. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Bonanni D, Citarella A, Moi D, Pinzi L, Bergamini E and Rastelli G: Dual Targeting strategies on histone deacetylase 6 (HDAC6) and heat shock protein 90 (Hsp90). Curr Med Chem. 29:1474–1502. 2022. View Article : Google Scholar | |
|
Ryu HW, Won HR, Lee DH and Kwon SH: HDAC6 regulates sensitivity to cell death in response to stress and post-stress recovery. Cell Stress Chaperones. 22:253–261. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Barter MJ, Butcher A, Wang H, Tsompani D, Galler M, Rumsby EL, Culley KL, Clark IM and Young DA: HDAC6 regulates NF-κB signalling to control chondrocyte IL-1-induced MMP and inflammatory gene expression. Sci Rep. 12:66402022. View Article : Google Scholar | |
|
Zhang WB, Yang F, Wang Y, Jiao FZ, Zhang HY, Wang LW and Gong ZJ: Inhibition of HDAC6 attenuates LPS-induced inflammation in macrophages by regulating oxidative stress and suppressing the TLR4-MAPK/NF-κB pathways. Biomed Pharmacother. 117:1091662019. View Article : Google Scholar | |
|
Xu S, Chen H, Ni H and Dai Q: Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-κB/NLRP3 pathway. Atherosclerosis. 317:1–9. 2021. View Article : Google Scholar | |
|
Zhang J, Liu M, Liu W and Wang W: Ras-ERK1/2 signalling promotes the development of osteosarcoma through regulation of H4K12ac through HAT1. Artif Cells Nanomed Biotechnol. 47:1207–1215. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Huang Z, Xia Y, Hu K, Zeng S, Wu L, Liu S, Zhi C, Lai M, Chen D, Xie L and Yuan Z: Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J Neurochem. 152:221–234. 2020. View Article : Google Scholar | |
|
Lv R, Liu X, Zhang Y, Dong N, Wang X, He Y, Yue H and Yin Q: Pathophysiological mechanisms and therapeutic approaches in obstructive sleep apnea syndrome. Signal Transduct Target Ther. 8:2182023. View Article : Google Scholar : PubMed/NCBI | |
|
Ou Y, Shen C, Chen Z, Liu T, Peng Y, Zong D and Ouyang R: TDP43/HDAC6/Prdx1 signaling pathway participated in the cognitive impairment of obstructive sleep apnea via regulating inflammation and oxidative stress. Int Immunopharmacol. 127:1113502024. View Article : Google Scholar | |
|
Tolosa E, Garrido A, Scholz SW and Poewe W: Challenges in the diagnosis of Parkinson's disease. Lancet Neurol. 20:385–397. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al: Parkin and PINK1 mitigate STING-induced inflammation. Nature. 561:258–262. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Yan S, Wei X, Jian W, Qin Y, Liu J, Zhu S, Jiang F, Lou H and Zhang B: Pharmacological inhibition of HDAC6 attenuates NLRP3 inflammatory response and protects dopaminergic neurons in experimental models of Parkinson's disease. Front Aging Neurosci. 12:782020. View Article : Google Scholar : PubMed/NCBI | |
|
Peng C, Wang Y, Hu Z and Chen C: Selective HDAC6 inhibition protects against blood-brain barrier dysfunction after intracerebral hemorrhage. CNS Neurosci Ther. 30:e144292024. View Article : Google Scholar | |
|
Wang M, Zhou C, Yu L, Kong D, Ma W, Lv B, Wang Y, Wu W, Zhou M and Cui G: Upregulation of MDH1 acetylation by HDAC6 inhibition protects against oxidative stress-derived neuronal apoptosis following intracerebral hemorrhage. Cell Mol Life Sci. 79:3562022. View Article : Google Scholar : PubMed/NCBI | |
|
Gómez-Benito M, Granado N, Garcia-Sanz P, Michel A, Dumoulin M and Moratalla R: Modeling Parkinson's disease with the alpha-synuclein protein. Front Pharmacol. 11:3562020. View Article : Google Scholar : PubMed/NCBI | |
|
Masini D, Plewnia C, Bertho M, Scalbert N, Caggiano V and Fisone G: A guide to the generation of a 6-hydroxydopamine mouse model of Parkinson's disease for the study of non-motor symptoms. Biomedicines. 9:5982021. View Article : Google Scholar : PubMed/NCBI | |
|
Bobrowska A, Paganetti P, Matthias P and Bates GP: Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington's disease. PLoS One. 6:e206962011. View Article : Google Scholar : PubMed/NCBI | |
|
Mazzetti S, De Leonardis M, Gagliardi G, Calogero AM, Basellini MJ, Madaschi L, Costa I, Cacciatore F, Spinello S, Bramerio M, et al: Phospho-HDAC6 gathers into protein aggregates in Parkinson's disease and atypical parkinsonisms. Front Neurosci. 14:6242020. View Article : Google Scholar : PubMed/NCBI | |
|
D'Sa K, Evans JR, Virdi GS, Vecchi G, Adam A, Bertolli O, Fleming J, Chang H, Leighton C, Horrocks MH, et al: Prediction of mechanistic subtypes of Parkinson's using patient-derived stem cell models. Nat Mach Intell. 5:933–946. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Marek K, Chowdhury S, Siderowf A, Lasch S, Coffey CS, Caspell-Garcia C, Simuni T, Jennings D, Tanner CM, Trojanowski JQ, et al: The Parkinson's progression markers initiative (PPMI)-establishing a PD biomarker cohort. Ann Clin Transl Neurol. 5:1460–1477. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Cheng T, Liu C, Wang Y, Li G, Feng L, Zhang S, Qi B, Cui J, Guo L, Cao L, et al: A novel histone deacetylase inhibitor Se-SAHA attenuates isoproterenol-induced heart failure via antioxidative stress and autophagy inhibition. Toxicol Appl Pharmacol. 487:1169572024. View Article : Google Scholar : PubMed/NCBI | |
|
Ruopp NF and Cockrill BA: Diagnosis and treatment of pulmonary arterial hypertension: A review. JAMA. 327:1379–1391. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Chakraborty P, Po SS, Scherlag BJ and Dasari TW: The neurometabolic axis: A novel therapeutic target in heart failure. Life Sci. 333:1221222023. View Article : Google Scholar : PubMed/NCBI | |
|
Brandt M, Dörschmann H, Khraisat S, Knopp T, Ringen J, Kalinovic S, Garlapati V, Siemer S, Molitor M, Göbel S, et al: Telomere shortening in hypertensive heart disease depends on oxidative DNA damage and predicts impaired recovery of cardiac function in heart failure. Hypertension. 79:2173–2184. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao Z, Liu J, Hu Y, Zhang X, Cao L, Dong Z, Li L and Hu Z: Bacterial diversity in the intestinal mucosa of heart failure rats treated with Sini Decoction. BMC Complement Med Ther. 22:932022. View Article : Google Scholar : PubMed/NCBI | |
|
Li C, Chang J, Wang Y and Pan G: Indole-3-propionic acid, a product of intestinal flora, inhibits the HDAC6/NOX2 signalling and relieves doxorubicin-induced cardiomyocyte damage. Folia Morphol (Warsz). 83:382–390. 2024. | |
|
De Becker B and Van De Borne P: Serum uric acid: A futile bystander in endothelial function? Blood Press. 32:22371232023. View Article : Google Scholar : PubMed/NCBI | |
|
Yu W and Cheng JD: Uric acid and cardiovascular disease: An update from molecular mechanism to clinical perspective. Front Pharmacol. 11:5826802020. View Article : Google Scholar : PubMed/NCBI | |
|
Wang K, Zhang Y, Zhou M, Du Y, Li P, Guan C and Huang Z: HDAC inhibitors alleviate uric acid-induced vascular endothelial cell injury by way of the HDAC6/FGF21/PI3K/AKT pathway. J Cardiovasc Pharmacol. 81:150–164. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Guymer RH and Campbell TG: Age-related macular degeneration. Lancet. 401:1459–1472. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Fleckenstein M, Keenan TDL, Guymer RH, Chakravarthy U, Schmitz-Valckenberg S, Klaver CC, Wong WT and Chew EY: Age-related macular degeneration. Nat Rev Dis Primers. 7:312021. View Article : Google Scholar : PubMed/NCBI | |
|
Fleckenstein M, Schmitz-Valckenberg S and Chakravarthy U: Age-related macular degeneration: A review. JAMA. 331:147–157. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Sundaramurthi H, Roche SL, Grice GL, Moran A, Dillion ET, Campiani G, Nathan JA and Kennedy BN: Selective histone deacetylase 6 inhibitors restore cone photoreceptor vision or outer segment morphology in zebrafish and mouse models of retinal blindness. Front Cell Dev Biol. 8:6892020. View Article : Google Scholar : PubMed/NCBI | |
|
Abouhish H, Thounaojam MC, Jadeja RN, Gutsaeva DR, Powell FL, Khriza M, Martin PM and Bartoli M: Inhibition of HDAC6 attenuates diabetes-induced retinal redox imbalance and microangiopathy. Antioxidants (Basel). 9. pp. 5992020, View Article : Google Scholar | |
|
Yang Q, Li S, Zhou Z, Fu M, Yang X, Hao K and Liu Y: HDAC6 inhibitor Cay10603 inhibits high glucose-induced oxidative stress, inflammation and apoptosis in retinal pigment epithelial cells via regulating NF-κB and NLRP3 inflammasome pathway. Gen Physiol Biophys. 39:169–177. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Hou Q, Kan S, Wang Z, Shi J, Zeng C, Yang D, Jiang S and Liu Z: Inhibition of HDAC6 with CAY10603 ameliorates diabetic kidney disease by suppressing NLRP3 inflammasome. Front Pharmacol. 13:9383912022. View Article : Google Scholar : PubMed/NCBI | |
|
Zeng Y, Wu R, Wang F, Li S, Li L, Li Y, Qin P, Wei M, Yang J, Wu J, et al: Liberation of daidzein by gut microbial β-galactosidase suppresses acetaminophen-induced hepatotoxicity in mice. Cell Host Microbe. 31:766–780.e7. 2023. View Article : Google Scholar | |
|
Chen S, Lu Z, Jia H, Yang B, Liu C, Yang Y, Zhang S, Wang Z, Yang L, Li S, et al: Hepatocyte-specific Mas activation enhances lipophagy and fatty acid oxidation to protect against acetaminophen-induced hepatotoxicity in mice. J Hepatol. 78:543–557. 2023. View Article : Google Scholar | |
|
Jaeschke H and Ramachandran A: Acetaminophen hepatotoxicity: Paradigm for understanding mechanisms of drug-induced liver injury. Annu Rev Pathol. 19:453–478. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang GD, Wang LL, Zheng L, Wang SQ, Yang RQ, He YT, Wang JW, Zhao MY, Ding Y, Liu M, et al: A novel HDAC6 inhibitor attenuate APAP-induced liver injury by regulating MDH1-mediated oxidative stress. Int Immunopharmacol. 131:1118612024. View Article : Google Scholar : PubMed/NCBI | |
|
Cecconi M, Evans L, Levy M and Rhodes A: Sepsis and septic shock. Lancet. 392:75–87. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Liu D, Huang SY, Sun JH, Zhang HC, Cai QL, Gao C, Li L, Cao J, Xu F, Zhou Y, et al: Sepsis-induced immunosuppression: Mechanisms, diagnosis and current treatment options. Mil Med Res. 9:562022.PubMed/NCBI | |
|
Vincent JL: Current sepsis therapeutics. EBioMedicine. 86:1043182022. View Article : Google Scholar : PubMed/NCBI | |
|
Guo SD, Yan ST, Li W, Zhou H, Yang JP, Yao Y, Shen MJ, Zhang LW, Zhang HB and Sun LC: HDAC6 promotes sepsis development by impairing PHB1-mediated mitochondrial respiratory chain function. Aging (Albany NY). 12:5411–5422. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Capuzzimati M, Hough O and Liu M: Cell death and ischemia-reperfusion injury in lung transplantation. J Heart Lung Transplant. 41:1003–1013. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Wang YH, Yan ZZ, Luo SD, Hu JJ, Wu M, Zhao J, Liu WF, Li C and Liu KX: Gut microbiota-derived succinate aggravates acute lung injury after intestinal ischaemia/reperfusion in mice. Eur Respir J. 61:22008402023. View Article : Google Scholar | |
|
Zhou H, Wu R and Li H: Downregulation of HDAC6 mitigates lung ischemia/reperfusion injury depending on activation of Nrf2/HO-1 signaling pathway and inactivation of ERK/NF-κB signaling pathway. Tissue Cell. 89:1024462024. View Article : Google Scholar | |
|
Abramoff B and Caldera FE: Osteoarthritis: Pathology, diagnosis, and treatment options. Med Clin North Am. 104:293–311. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Favero M, Belluzzi E, Ortolan A, Lorenzin M, Oliviero F, Doria A, Scanzello CR and Ramonda R: Erosive hand osteoarthritis: Latest findings and outlook. Nat Rev Rheumatol. 18:171–183. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Knights AJ, Redding SJ and Maerz T: Inflammation in osteoarthritis: The latest progress and ongoing challenges. Curr Opin Rheumatol. 35:128–134. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Shen Z, Ji K, Cai Z, Huang C, He X, Xu H and Chen G: Inhibition of HDAC6 by Tubastatin A reduces chondrocyte oxidative stress in chondrocytes and ameliorates mouse osteoarthritis by activating autophagy. Aging (Albany NY). 13:9820–9837. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Cope PJ, Ourradi K, Li Y and Sharif M: Models of osteoarthritis: The good, the bad and the promising. Osteoarthritis Cartilage. 27:230–239. 2019. View Article : Google Scholar : | |
|
Malfait AM and Little CB: On the predictive utility of animal models of osteoarthritis. Arthritis Res Ther. 17:2252015. View Article : Google Scholar : PubMed/NCBI | |
|
Teng M, Zhao X, Wang C, Wang C, White JC, Zhao W, Zhou L, Duan M and Wu F: Polystyrene nanoplastics toxicity to zebrafish: Dysregulation of the brain-intestine-microbiota axis. ACS Nano. 16:8190–8204. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Shan S, Zhang Y, Zhao H, Zeng T and Zhao X: Polystyrene nanoplastics penetrate across the blood-brain barrier and induce activation of microglia in the brain of mice. Chemosphere. 298:1342612022. View Article : Google Scholar : PubMed/NCBI | |
|
Wang B, Liang B, Huang Y, Li Z, Zhang B, Du J, Ye R, Xian H, Deng Y, Xiu J, et al: Long-chain Acyl carnitines aggravate polystyrene nanoplastics-induced atherosclerosis by upregulating MARCO. Adv Sci (Weinh). 10:e22058762023. View Article : Google Scholar : PubMed/NCBI | |
|
Qiu W, Ye J, Su Y, Zhang X, Pang X, Liao J, Wang R, Zhao C, Zhang H, Hu L, et al: Co-exposure to environmentally relevant concentrations of cadmium and polystyrene nanoplastics induced oxidative stress, ferroptosis and excessive mitophagy in mice kidney. Environ Pollut. 333:1219472023. View Article : Google Scholar : PubMed/NCBI | |
|
Han SW and Ryu KY: Increased clearance of non-biodegradable polystyrene nanoplastics by exocytosis through inhibition of retrograde intracellular transport. J Hazard Mater. 439:1295762022. View Article : Google Scholar : PubMed/NCBI | |
|
Fan T, Wang X, Zhang S, Deng P, Jiang Y, Liang Y, Jie S, Wang Q, Li C, Tian G, et al: NUPR1 promotes the proliferation and metastasis of oral squamous cell carcinoma cells by activating TFE3-dependent autophagy. Signal Transduct Target Ther. 7:1302022. View Article : Google Scholar : PubMed/NCBI | |
|
Hasegawa K, Fujii S, Matsumoto S, Tajiri Y, Kikuchi A and Kiyoshima T: YAP signaling induces PIEZO1 to promote oral squamous cell carcinoma cell proliferation. J Pathol. 253:80–93. 2021. View Article : Google Scholar | |
|
Bayik D and Lathia JD: Cancer stem cell-immune cell crosstalk in tumour progression. Nat Rev Cancer. 21:526–536. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Jin G, Wang K, Zhao Y, Yuan S, He Z and Zhang J: Targeting histone deacetylases for heart diseases. Bioorg Chem. 138:1066012023. View Article : Google Scholar : PubMed/NCBI | |
|
Tavares MO, Milan TM, Bighetti-Trevisan RL, Leopoldino AM and de Almeida LO: Pharmacological inhibition of HDAC6 overcomes cisplatin chemoresistance by targeting cancer stem cells in oral squamous cell carcinoma. J Oral Pathol Med. 51:529–537. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Jomova K, Raptova R, Alomar SY, Alwasel SH, Nepovimova E, Kuca K and Valko M: Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch Toxicol. 97:2499–2574. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Nagai H, Tatara H, Tanaka-Furuhashi K, Kurata S and Yano T: Homeostatic regulation of ROS-triggered Hippo-Yki pathway via autophagic clearance of Ref(2)P/p62 in the Drosophila intestine. Dev Cell. 56:81–94.e10. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Ando T, Arang N, Wang Z, Costea DE, Feng X, Goto Y, Izumi H, Gilardi M, Ando K and Gutkind JS: EGFR regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1. Commun Biol. 4:12372021. View Article : Google Scholar : PubMed/NCBI | |
|
Jin J, Zhang L, Li X, Xu W, Yang S, Song J, Zhang W, Zhan J, Luo J and Zhang H: Oxidative stress-CBP axis modulates MOB1 acetylation and activates the Hippo signaling pathway. Nucleic Acids Res. 50:3817–3834. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Cai R, Zhu H, Liu Y, Sha H, Peng W, Yin R, Zhou G and Fang Y: To be, or not to be: the dilemma of immunotherapy for non-small cell lung cancer harboring various driver mutations. J Cancer Res Clin Oncol. 149:10027–10040. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Bourouh M and Marignani PA: The tumor suppressor kinase LKB1: Metabolic nexus. Front Cell Dev Biol. 10:8812972022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang H, Nabel CS, Li D, O'Connor RÍ, Crosby CR, Chang SM, Hao Y, Stanley R, Sahu S, Levin DS, et al: Histone deacetylase 6 inhibition exploits selective metabolic vulnerabilities in LKB1 mutant, KRAS driven NSCLC. J Thorac Oncol. 18:882–895. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wißmann N, Botz M, Soyka SJ, Beretta CA, Pramatarov RL, et al: Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell. 185:2899–2917.e31. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Schaff LR and Mellinghoff IK: Glioblastoma and other primary brain malignancies in adults: A review. JAMA. 329:574–587. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Sharma R, Chiang YH, Chen HC, Lin HY, Yang WB, Nepali K, Lai MJ, Chen KY, Liou JP and Hsu TI: Dual inhibition of CYP17A1 and HDAC6 by abiraterone-installed hydroxamic acid overcomes temozolomide resistance in glioblastoma through inducing DNA damage and oxidative stress. Cancer Lett. 586:2166662024. View Article : Google Scholar : PubMed/NCBI | |
|
Yan X, Qu X, Liu B, Zhao Y, Xu L, Yu S, Wang J, Wang L and Su J: Autophagy-induced HDAC6 activity during hypoxia regulates mitochondrial energy metabolism through the β-catenin/COUP-TFII axis in hepatocellular carcinoma cells. Front Oncol. 11:7424602021. View Article : Google Scholar | |
|
Zeleke TZ, Pan Q, Chiuzan C, Onishi M, Li Y, Tan H, Alvarez MJ, Honan E, Yang M, Chia PL, et al: Network-based assessment of HDAC6 activity predicts preclinical and clinical responses to the HDAC6 inhibitor ricolinostat in breast cancer. Nat Cancer. 4:257–275. 2023. View Article : Google Scholar | |
|
Xu G, Niu L, Wang Y, Yang G, Zhu X, Yao Y, Zhao G, Wang S and Li H: HDAC6-dependent deacetylation of TAK1 enhances sIL-6R release to promote macrophage M2 polarization in colon cancer. Cell Death Dis. 13:8882022. View Article : Google Scholar : PubMed/NCBI | |
|
Mardones C, Navarrete-Munoz C, Armijo ME, Salgado K, Rivas-Valdes F, Gonzalez-Pecchi V, Farkas C, Villagra A and Hepp MI: Role of HDAC6-STAT3 in immunomodulatory pathways in colorectal cancer cells. Mol Immunol. 164:98–111. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Jiang D and Ma P: Canagliflozin, characterized as a HDAC6 inhibitor, inhibits gastric cancer metastasis. Front Oncol. 12:10574552022. View Article : Google Scholar : PubMed/NCBI | |
|
Zheng Y and Yang X, Wang C, Zhang S, Wang Z, Li M, Wang Y, Wang X and Yang X: HDAC6, modulated by miR-206, promotes endometrial cancer progression through the PTEN/AKT/mTOR pathway. Sci Rep. 10:35762020. View Article : Google Scholar : PubMed/NCBI | |
|
Dai HY, Chang LS, Yang SF, Wang SN, Su SJ and Yeh YT: HDAC6 promotes aggressive development of liver cancer by improving egfr mRNA stability. Neoplasia. 35:1008452023. View Article : Google Scholar | |
|
Guadagni A, Barone S, Alfano AI, Pelliccia S, Bello I, Panza E, Summa V and Brindisi M: Tackling triple negative breast cancer with HDAC inhibitors: 6 is the isoform! Eur J Med Chem. 279:1168842024. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang SL, Zhu HY, Zhou BY, Chu Y, Huo JR, Tan YY and Liu DL: Histone deacetylase 6 is overexpressed and promotes tumor growth of colon cancer through regulation of the MAPK/ERK signal pathway. Onco Targets Ther. 12:2409–2419. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou B, Liu D and Tan Y: Role of HDAC6 and its selective inhibitors in gastrointestinal cancer. Front Cell Dev Biol. 9:7193902021. View Article : Google Scholar : PubMed/NCBI | |
|
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a microtubule-associated deacetylase. Nature. 417:455–458. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Kovacs JJ, Murphy PJM, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB and Yao TP: HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 18:601–607. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, et al: HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 27:197–213. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Yoo J, Jeon YH, Lee DH, Kim GW, Lee SW, Kim SY, Park J and Kwon SH: HDAC6-selective inhibitors enhance anticancer effects of paclitaxel in ovarian cancer cells. Oncol Lett. 21:2012021. View Article : Google Scholar : PubMed/NCBI | |
|
Bridgewater JA, Beer P, Bendall J, Dehbi HM, Duggan M, Hung M, King J, McElwaine-Johnn H and White L: A phase II study of selective HDAC6 inhibition with KA2507 for second-line treatment of advanced biliary tract cancer (ABC-11). J Clin Oncol. 38(15 Suppl): TPS46522020. View Article : Google Scholar | |
|
Park SJ, Joo SH, Lee N, Jang WJ, Seo JH and Jeong CH: ACY-241, an HDAC6 inhibitor, overcomes erlotinib resistance in human pancreatic cancer cells by inducing autophagy. Arch Pharm Res. 44:1062–1075. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Shao Y, Zhu F, Zhu S and Bai L: HDAC6 suppresses microRNA-199a transcription and augments HPV-positive cervical cancer progression through Wnt5a upregulation. Int J Biochem Cell Biol. 136:1060002021. View Article : Google Scholar : PubMed/NCBI | |
|
Zuo Q, Wu W, Li X, Zhao L and Chen W: HDAC6 and SIRT2 promote bladder cancer cell migration and invasion by targeting cortactin. Oncol Rep. 27:819–824. 2012. | |
|
Ruan Y, Wang L and Lu Y: HDAC6 inhibitor, ACY1215 suppress the proliferation and induce apoptosis of gallbladder cancer cells and increased the chemotherapy effect of gemcitabine and oxaliplatin. Drug Dev Res. 82:598–604. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Yang Y, Sun Y, Wang H, Li H, Zhang M, Zhou L, Meng X, Wu Y, Liu P, Liu X, et al: MicroRNA-221 induces autophagy through suppressing HDAC6 expression and promoting apoptosis in pancreatic cancer. Oncol Lett. 16:7295–7301. 2018.PubMed/NCBI | |
|
Kim JY, Han SY, Yoo J, Kim GW, Jeon YH, Lee SW, Park J and Kwon SH: HDAC8-selective inhibition by PCI-34051 enhances the anticancer effects of ACY-241 in ovarian cancer cells. Int J Mol Sci. 23:86452022. View Article : Google Scholar : PubMed/NCBI | |
|
Nawar N, Bukhari S, Adile AA, Suk Y, Manaswiyoungkul P, Toutah K, Olaoye OO, Raouf YS, Sedighi A, Garcha HK, et al: Discovery of HDAC6-selective inhibitor NN-390 with in vitro efficacy in group 3 medulloblastoma. J Med Chem. 65:3193–3217. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Sun J, Wu W, Tang X, Zhang F, Ju C, Liu R, Liang Y, Yu B, Lv B, Guo Y, et al: HDAC6 inhibitor WT161 performs anti-tumor effect on osteosarcoma and synergistically interacts with 5-FU. Biosci Rep. 41:BSR202039052021. View Article : Google Scholar : PubMed/NCBI | |
|
Garcha HK, Nawar N, Sorger H, Erdogan F, Aung MMK, Sedighi A, Manaswiyoungkul P, Seo HS, Schönefeldt S, Pölöske D, et al: High efficacy and drug synergy of HDAC6-selective inhibitor NN-429 in natural killer (NK)/T-cell lymphoma. Pharmaceuticals (Basel). 15. pp. 13212022, View Article : Google Scholar | |
|
Zhang J, Chen X, Chen G, Wang H, Jia L, Hao Y and Yao D: Identification of a novel PAK1/HDAC6 dual inhibitor ZMF-23 that triggers tubulin-stathmin regulated cell death in triple negative breast cancer. Int J Biol Macromol. 251:1263482023. View Article : Google Scholar : PubMed/NCBI | |
|
Reddy RG, Surineni G, Bhattacharya D, Marvadi SK, Sagar A, Kalle AM, Kumar A, Kantevari S and Chakravarty S: Crafting carbazole-based Vorinostat and tubastatin-A-like histone deacetylase (HDAC) inhibitors with potent in vitro and in vivo neuroactive functions. ACS Omega. 4:17279–17294. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Leyk J, Daly C, Janssen-Bienhold U, Kennedy BN and Richter-Landsberg C: HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 8:e30282017. View Article : Google Scholar : PubMed/NCBI | |
|
Chen J, Zhang J, Shaik NF, Yi B, Wei X, Yang XF, Naik UP, Summer R, Yan G, Xu X and Sun J: The histone deacetylase inhibitor tubacin mitigates endothelial dysfunction by up-regulating the expression of endothelial nitric oxide synthase. J Biol Chem. 294:19565–19576. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Nomura Y, Nakano M, Woo Sung H, Han M and Pandey D: Inhibition of HDAC6 activity protects against endothelial dysfunction and atherogenesis in vivo: A role for HDAC6 neddylation. Front Physiol. 12:6757242021. View Article : Google Scholar : PubMed/NCBI | |
|
Wang N, Wang H, Chen J, Wang F, Wang S, Zhou Q, Ying J, Huang S, Wang P and Yuan F: ACY-1215, a HDAC6 inhibitor, decreases the dexamethasone-induced suppression of osteogenesis in MC3T3-E1 cells. Mol Med Rep. 22:2451–2459. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Chen C, Liu A, Lu Q, Luo L, Li J, Ke J, Liu Y and Feng X: HDAC6 inhibitor ACY-1215 improves neuropathic pain and its comorbidities in rats of peripheral nerve injury by regulating neuroinflammation. Chem Biol Interact. 353:1098032022. View Article : Google Scholar : PubMed/NCBI | |
|
Yan B, Xie S, Liu Z, Ran J, Li Y, Wang J, Yang Y, Zhou J, Li D and Liu M: HDAC6 deacetylase activity is critical for lipopolysaccharide-induced activation of macrophages. PLoS One. 9:e1107182014. View Article : Google Scholar : PubMed/NCBI | |
|
Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, Whittaker S, Tokura Y, Vermeer M, Zinzani PL, et al: Mogamulizumab versus Vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): An international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 19:1192–1204. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Guo C, Wang Q, Zhang X, Lu F, Sun M, Zeng P, Sun L, She L, Wang B, Zhang Y, et al: Gelated Vorinostat with inner-lysosome triggered release for tumor-targeting chemotherapy. Colloids Surf B Biointerfaces. 194:1111442020. View Article : Google Scholar : PubMed/NCBI | |
|
Praseetha S, Bandaru S, Yadav M, Nayarisseri A and Sureshkumar S: Common SAR derived from multiple QSAR models on vorinostat derivatives targeting HDACs in tumor treatment. Curr Pharm Des. 22:5072–5078. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Wang Z, Leng Y, Wang J, Liao HM, Bergman J, Leeds P, Kozikowski A and Chuang DM: Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: Potential roles of α-tubulin acetylation and FGF-21 up-regulation. Sci Rep. 6:196262016. View Article : Google Scholar | |
|
Shen S, Svoboda M, Zhang G, Cavasin MA, Motlova L, McKinsey TA, Eubanks JH, Bařinka C and Kozikowski AP: Structural and in vivo characterization of tubastatin A, a widely used histone deacetylase 6 inhibitor. ACS Med Chem Lett. 11:706–712. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Cosenza M and Pozzi S: The therapeutic strategy of HDAC6 inhibitors in lymphoproliferative disease. Int J Mol Sci. 19:23372018. View Article : Google Scholar : PubMed/NCBI | |
|
Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, Orlowski R, Supko JG, Tamang D, Yang M, Jones SS, et al: Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res. 23:3307–3315. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Yee AJ, Bensinger WI, Supko JG, Voorhees PM, Berdeja JG, Richardson PG, Libby EN, Wallace EE, Birrer NE, Burke JN, et al: Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 17:1569–1578. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Adeegbe DO, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, Dries R, Li Y, Liu S, Wang X, et al: Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov. 7:852–867. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Hideshima T, Qi J, Paranal RM, Tang W, Greenberg E, West N, Colling ME, Estiu G, Mazitschek R, Perry JA, et al: Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc Natl Acad Sci USA. 113:13162–13167. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang QQ, Zhang WJ and Chang S: HDAC6 inhibition: A significant potential regulator and therapeutic option to translate into clinical practice in renal transplantation. Front Immunol. 14:11688482023. View Article : Google Scholar : PubMed/NCBI | |
|
Granzotto A, Vissel B and Sensi SL: Lost in translation: Inconvenient truths on the utility of mouse models in Alzheimer's disease research. Elife. 13:e906332024. View Article : Google Scholar : PubMed/NCBI | |
|
Dovonou A, Bolduc C, Soto Linan V, Gora C, Peralta Iii MR and Lévesque M: Animal models of Parkinson's disease: Bridging the gap between disease hallmarks and research questions. Transl Neurodegener. 12:362023. View Article : Google Scholar : PubMed/NCBI | |
|
Karp NA, Berdoy M, Gray K, Hunt L, Jennings M, Kerton A, Leach M, Tremoleda JL, Gledhill J, Pearl EJ, et al: The sex inclusive research framework to address sex bias in preclinical research proposals. Nat Commun. 16:37632025. View Article : Google Scholar : PubMed/NCBI | |
|
Li Y, Sang S, Ren W, Pei Y, Bian Y, Chen Y and Sun H: Inhibition of histone deacetylase 6 (HDAC6) as a therapeutic strategy for Alzheimer's disease: A review (2010-2020). Eur J Med Chem. 226:1138742021. View Article : Google Scholar : PubMed/NCBI | |
|
Phipps AJ, Dwyer S, Collins JM, Kabir F, Atkinson RA, Chowdhury MA, Matthews L, Dixit D, Terry RS, Smith J, et al: HDAC6 inhibition as a mechanism to prevent neurodegeneration in the mSOD1G93A mouse model of ALS. Heliyon. 10:e345872024. View Article : Google Scholar : | |
|
Takizawa D, Kakizaki S, Horiguchi N, Tojima H, Yamazaki Y, Ichikawa T, Sato K and Mori M: Histone deacetylase inhibitors induce cytochrome P450 2B by activating nuclear receptor constitutive androstane receptor. Drug Metab Dispos. 38:1493–1498. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et al: Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 119:2579–2589. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Huang Z, Li L, Cheng B and Li D: Small molecules targeting HDAC6 for cancer treatment: Current progress and novel strategies. Biomed Pharmacother. 178:1172182024. View Article : Google Scholar : PubMed/NCBI | |
|
Regna NL, Vieson MD, Luo XM, Chafin CB, Puthiyaveetil AG, Hammond SE, Caudell DL, Jarpe MB and Reilly CM: Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin Immunol. 162:58–73. 2016. View Article : Google Scholar |










