



Harnessing CRISPR/Cas9 to overcome targeted therapy resistance in non‑small cell lung cancer: Advances and challenges (Review)
- Authors:
- Published online on: July 9, 2025 https://doi.org/10.3892/or.2025.8944
- Article Number: 111
-
Copyright: © Du et al. This is an open access article distributed under the terms of Creative Commons Attribution License [CC BY_NC 4.0].
Abstract
Introduction
Lung cancer is a highly malignant and heterogeneous disease and remains the leading cause of cancer-related mortality worldwide (1). It is broadly classified into two histological subtypes: Small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), with NSCLC accounting for ~85% of all cases (2). Despite significant advances in diagnostics and therapeutics, the prognosis for patients with advanced NSCLC remains poor, with a five-year survival rate <15%, particularly in those with metastatic disease (1,2). Traditional chemotherapy and radiotherapy have shown limited efficacy, underscoring the urgent need for more effective and targeted treatment strategies.
The discovery of oncogenic driver mutations has revolutionized the treatment landscape of NSCLC. Targeted therapies aimed at these genetic alterations, such as EGFR, ALK, KRAS and others, have demonstrated remarkable clinical benefits in subsets of patients, improving response rates and progression-free survival (3,4). However, the development of acquired resistance to these therapies remains a major therapeutic hurdle, often leading to disease progression and limited long-term outcomes (5). Therefore, there is an urgent need for new treatment strategies to improve the response to targeted therapy.
The emergence of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene editing technology has provided powerful new tools for exploring the molecular mechanisms of drug resistance. Since the discovery of the CRISPR locus in 1987 and Cas genes in 2002, gene editing has rapidly evolved, with the CRISPR/Cas9 system becoming one of the most widely used platforms due to its simplicity, specificity and efficiency (6–9). This technology enables precise manipulation of genetic elements, allowing researchers to model resistance mechanisms, validate potential therapeutic targets and screen for synergistic drug combinations.
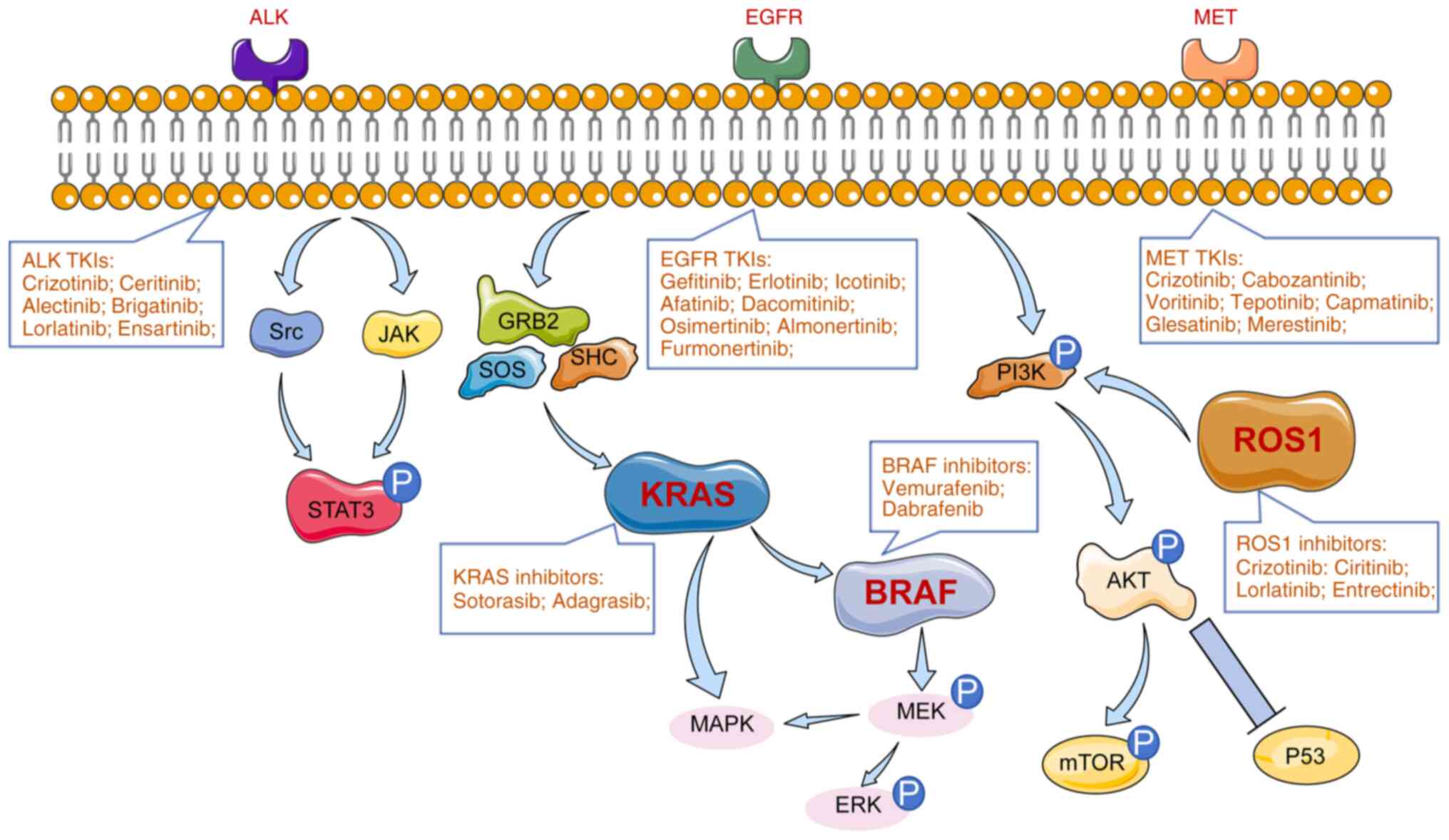
The present review focused on the role of CRISPR/Cas9 in elucidating the mechanisms of targeted therapy resistance in NSCLC. Specifically, it examined how this technology has been applied to study resistance associated with key driver genes such as EGFR, KRAS, ALK, ROS1, MET and BRAF (Fig. 1). By highlighting these advances, the present review aimed to provide a comprehensive overview of how CRISPR/Cas9 is shaping future strategies to overcome resistance and improve clinical outcomes in NSCLC.
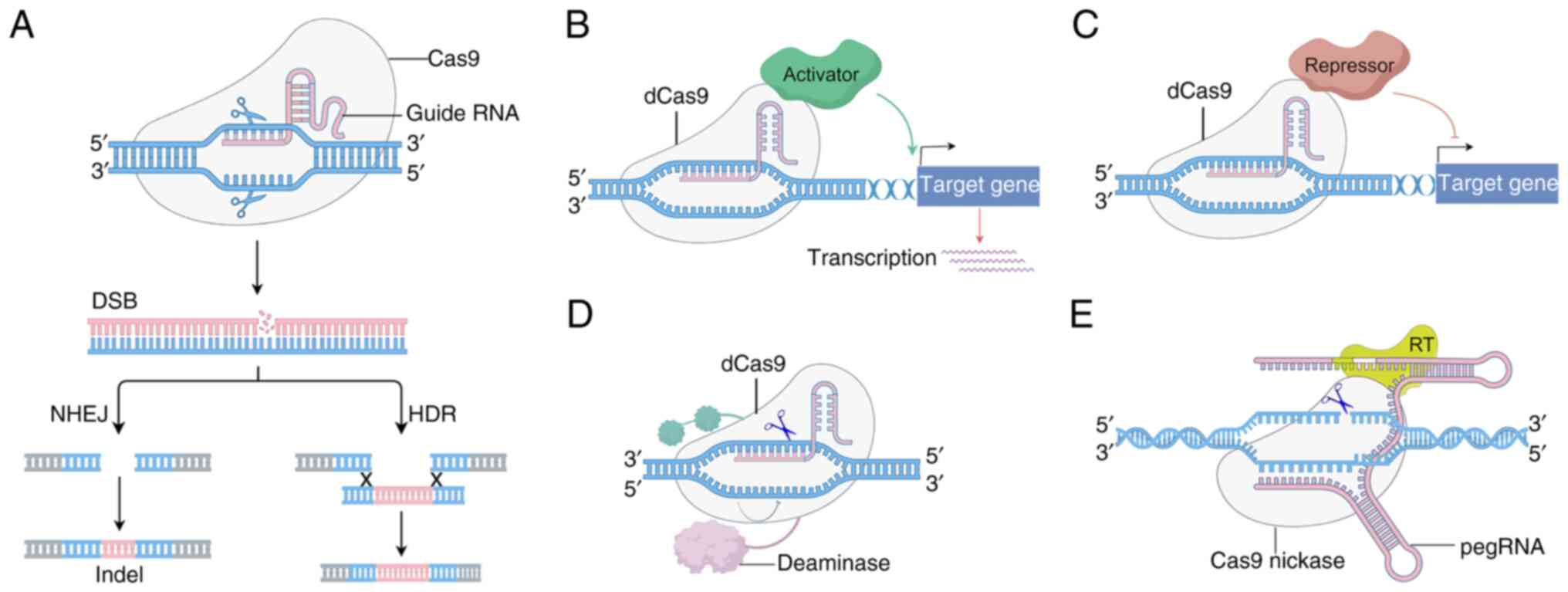
Overview of CRISPR/Cas9 technology
Basic principles of CRISPR/Cas9
The CRISPR/Cas9 system, derived from the adaptive immune system of bacteria, is a powerful genome-editing tool composed of two key components: The Cas9 endonuclease and a guide RNA (gRNA). The gRNA, comprising a CRISPR RNA (crRNA) and a trans-activating CRISPR RNA (tracrRNA), directs Cas9 to a specific genomic sequence through complementary base pairing (6–8). Upon recognition of the target DNA adjacent to a protospacer adjacent motif (PAM), the Cas9 protein induces a double-strand break (DSB). Cells repair these DSBs through one of two major pathways: non-homologous end joining, which is error-prone and may result in insertions or deletions (indels), or homology-directed repair, which uses a homologous template to accurately repair the break. This dual repair mechanism allows researchers to either disrupt gene function or introduce precise genetic modifications (Fig. 2). The programmability, efficiency and simplicity of CRISPR/Cas9 have made it a cornerstone in both basic research and therapeutic development. Its ability to target virtually any locus in the genome with high precision marks a significant advancement over previous editing technologies (9–11).
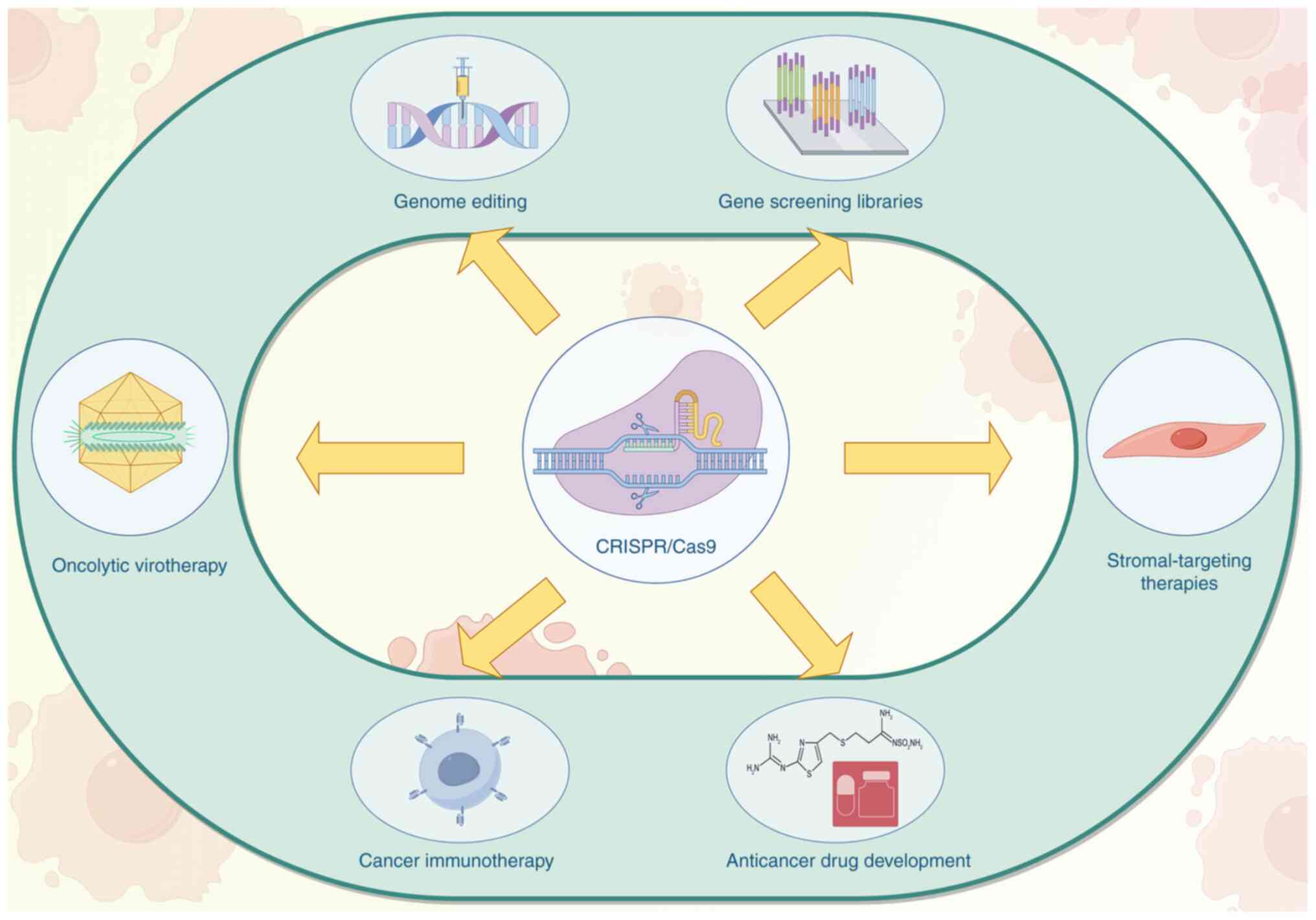
Applications of CRISPR/Cas9 in tumor research
CRISPR/Cas9 has transformed cancer research by enabling the functional interrogation of oncogenes and tumor suppressor genes, modeling of tumor evolution and developing treatment strategies (Fig. 3) (12–14). In NSCLC, it has facilitated the creation of genetically engineered cell lines and animal models that mirror specific mutations found in patients, such as EGFR, KRAS, or ALK alterations. These models are critical for studying tumorigenesis, drug response and resistance mechanisms. Moreover, CRISPR/Cas9 is widely used in high-throughput genetic screening to identify genes that mediate resistance or sensitivity to targeted therapies (15–17). These screens can uncover novel synthetic lethal interactions and inform rational combination therapies. As a result, CRISPR-based approaches are becoming indispensable in efforts to overcome therapy resistance in NSCLC and other malignancies.
Comparison with other gene editing technologies
Compared with earlier gene-editing platforms such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), CRISPR/Cas9 offers several advantages (18). It is easier to design, as it relies on RNA-based targeting rather than engineered protein-DNA interactions. This design flexibility accelerates experimentation and reduces cost (19). CRISPR/Cas9 also supports multiplex editing, targeting multiple genes simultaneously using different gRNAs, which is not easily achievable with ZFNs or TALENs (20). Despite its advantages, challenges remain, particularly with off-target effects, where the Cas9 nuclease may cut unintended genomic sites. However, the development of high-fidelity Cas9 variants and improved gRNA design algorithms have substantially mitigated these concerns (21–23). As the technology matures, CRISPR/Cas9 continues to expand its role from a basic research tool to a promising platform for clinical gene therapy, especially in oncology.
Studies on oncogenes in NSCLC
KRAS
The KRAS gene, a member of the RAS family of small GTPases, plays a critical role in cell proliferation, differentiation and survival. It is the most prevalent oncogenic driver mutation in NSCLC, observed in 20–30% of cases, with nearly 97% of mutations occurring as point mutations in codon 12 or 13 of exon 2 (24). These mutations result in constitutive activation of KRAS, leading to persistent downstream signaling through pathways such as MAPK and PI3K/AKT, ultimately promoting tumorigenesis and metastasis (25,26). The high prevalence of KRAS mutations in NSCLC underscores its importance as a therapeutic target and highlights the need for innovative strategies to inhibit its aberrant activity. However, the targeting of KRAS mutations in the treatment of lung cancer poses a significant challenge primarily due to the intricate structure and functionality of the KRAS protein, rendering it resistant to drug intervention. According to KRYSTAL-1 trial, 45% of patients developed resistances to KRAS inhibitor (27,28). Consequently, lung cancer patients harboring KRAS mutations experience limited treatment efficacy and the emergence of resistance is a prevalent concern (29). Consequently, patients with KRAS-mutated NSCLC have traditionally had limited treatment options and poor outcomes. However, recent advances have led to the development of covalent inhibitors specifically targeting the KRAS G12C mutation, a subset found in NSCLC. Notably, sotorasib (30) and adagrasib (28), demonstrated promising clinical efficacy and have been approved for use in KRAS G12C-mutated NSCLC. In a pivotal study, researchers employed a genome-scale CRISPR interference platform to systematically identify genetic vulnerabilities in KRAS G12C-mutant lung and pancreatic cancer models treated with KRAS G12C inhibitors (15). The study revealed ‘collateral dependencies’; genes whose loss increased cellular sensitivity to KRAS inhibition. These findings enabled the identification of two classes of combination therapies that either enhanced target engagement or inhibited compensatory survival pathways, offering a framework for rational therapeutic combinations to overcome resistance. Further advancing CRISPR applications, Gao et al (31) developed two CRISPR systems, CRISPR/SpCas9-mediated genome editing and a transcriptional repression system using dCas9-KRAB, to selectively target the KRAS G12S mutant allele. The study demonstrated effective suppression of mutant KRAS expression in A549 cells, leading to reduced phosphorylation of downstream effectors Akt and ERK, impaired tumor cell proliferation and significant tumor regression in vivo. Notably, the aforementioned analysis suggested that this mutation-specific editing strategy could be extended to a broader range of oncogenic mutations. Additionally, Dompe et al (32) used CRISPR screening to identify MAPK7 as a key therapeutic target in KRAS-mutant tumors. The authors' findings showed that combined inhibition of MAPK7 and MEK resulted in synergistic suppression of tumor growth in KRAS-mutant NSCLC xenograft models, highlighting the therapeutic potential of co-targeting downstream signaling nodes to overcome resistance. Together, these studies demonstrate the transformative potential of CRISPR/Cas9 technology in dissecting the molecular underpinnings of KRAS-driven NSCLC and in informing the development of novel targeted therapeutic strategies.
EGFR
The epidermal growth factor receptor (EGFR) is a transmembrane receptor tyrosine kinase that plays a central role in regulating cell proliferation, survival and differentiation. Activating mutations in EGFR are among the most common driver alterations in NSCLC, occurring in 10–20% of patients. These mutations, particularly exon 19 deletions and the L858R point mutation in exon 21, are associated with sensitivity to EGFR tyrosine kinase inhibitors (TKIs) (15,16,33). The introduction of first-generation EGFR-TKIs, such as gefitinib (34) and erlotinib (35), marked a paradigm shift in the treatment of advanced NSCLC. Subsequent generations of TKIs, including icotinib (36), afatinib (37), dacomitinib (38), osimertinib (39), almonertinib (40) and furmonertinib (41), further improved patient outcomes (Table I). Although EGFR-TKIs provide significant initial clinical benefits for patients with EGFR-mutated lung cancer, 50–60% of these patients eventually develop acquired resistance to first- or second-generation EGFR-TKIs. Moreover, 20–30% of EGFR-mutated patients exhibit primary resistance when treated with third-generation EGFR-TKIs as a first-line therapy (42–46). Common resistance mechanisms include secondary mutations in EGFR (such as T790M and C797S), activation of bypass pathways (such as MET amplification) and histological transformation to small cell lung cancer (47–50). The advent of CRISPR/Cas9 technology has provided a powerful platform to investigate these resistance mechanisms and develop strategies to overcome them. For example, Park et al (51) employed CRISPR/Cas9 to introduce the mTOR L1433S mutation into NSCLC cells, revealing that activation of the AKT signaling pathway may mediate resistance to third-generation EGFR-TKIs, such as osimertinib. The findings further demonstrated that dual inhibition of EGFR and mTOR signaling could restore drug sensitivity. Park et al (51) underscored the importance of CRISPR/Cas9 technology in uncovering innovative resistance mechanisms to EGFR TKIs. In another study, CRISPR/Cas9-mediated knockout of ZEB1 and FGFR1 identified their crucial roles in epithelial-mesenchymal transition (EMT)-associated resistance to EGFR-TKIs (52). Inhibition of ZEB1 impaired EMT processes and resensitized NSCLC cells to EGFR inhibitors, while targeting FGFR1 suppressed survival pathways activated during resistance development. Zeng et al (53) used genome-wide CRISPR/Cas9 screening to elucidate the role of RIC8A in modulating YAP pathway activity. Loss of RIC8A decreased the synergistic cytotoxic effects of EGFR-TKIs, highlighting potential targets for enhancing drug efficacy. Moreover, CRISPR/Cas9 was used to model the C797S mutation in EGFR, a key driver of resistance to third-generation TKIs. Functional studies revealed that overexpression of AXL contributed to reduced sensitivity to AZD9291 (osimertinib) and that AXL inhibition may represent a viable therapeutic strategy for C797S-mutant tumors (54). Finally, CRISPR/Cas9 barcoding technology has enabled tracking of heterogeneous subpopulations of NSCLC cells harboring specific resistance mutations (55). This approach allows dynamic modeling of resistance evolution under drug pressure and evaluation of combination therapies, offering important insights for optimizing treatment strategies. Collectively, these studies highlight the critical role of CRISPR/Cas9 in dissecting the complex molecular landscape of EGFR-TKI resistance in NSCLC and underscore its potential in identifying novel therapeutic vulnerabilities.
Anaplastic lymphoma kinase (ALK)
The ALK gene, located on chromosome 2p23, encodes a receptor tyrosine kinase that is part of the insulin receptor superfamily. In NSCLC, ALK gene rearrangements are found in 3–7% of cases and are considered key oncogenic drivers (56). Among the identified ALK fusion partners, EML4-ALK is the most common variant, accounting for ~85% of ALK-rearranged NSCLC cases (57). The fusion leads to constitutive activation of the ALK kinase domain, promoting uncontrolled cell proliferation and survival through downstream signaling pathways such as MAPK/ERK and PI3K/AKT (58,59). Crizotinib, the first ALK inhibitor, was approved by the FDA in 2011 and markedly improved outcomes for ALK-positive NSCLC patients (60–63). However, resistance to crizotinib and subsequent generations of ALK inhibitors, including ceritinib (64), alectinib (65), brigatinib (66) and lorlatinib (67) (Table II), commonly develops, limiting long-term efficacy (68). Previous studies have shown that ~40% of ALK-mutant patients develop resistance to ALK-TKIs, with ~10% exhibiting primary resistance and 30% showing acquired resistance (69,70). Resistance mechanisms to ALK-TKIs are diverse and include secondary mutations within the ALK kinase domain, gene amplification and activation of alternative signaling pathways such as EGFR, KIT and IGF1R (71,72). To investigate these mechanisms, researchers have employed CRISPR/Cas9-based genome editing to create precise models of ALK gene rearrangements in vitro (55). In a landmark study, Maddalo et al (73) used CRISPR/Cas9 to generate a mouse model harboring the EML4-ALK fusion, effectively recapitulating the genetic, histological and molecular features of ALK-rearranged NSCLC. These genetically engineered mice developed tumors that responded to ALK inhibitors, providing a robust platform for evaluating drug efficacy and resistance mechanisms in vivo. These genetically engineered mice developed tumors that responded to ALK inhibitors, providing a robust platform for evaluating drug efficacy and resistance mechanisms in vivo.
ROS1
The ROS1 gene, located on chromosome 6q22, encodes a receptor tyrosine kinase that belongs to the insulin receptor family. ROS1 rearrangements represent oncogenic fusion events in 1–2% of NSCLC cases and are considered actionable driver mutations (58–61). Due to structural homology between the kinase domains of ROS1 and ALK, several ALK inhibitors, including crizotinib, entrectinib, ceritinib and lorlatinib, have demonstrated efficacy against ROS1-rearranged NSCLC (74–81). While these targeted therapies provide significant initial clinical benefit, the emergence of acquired resistance remains a major barrier to long-term disease control. Resistance mechanisms include on-target mutations within the ROS1 kinase domain, such as G2032R and D2033N, that impair inhibitor binding, as well as less well-characterized off-target or bypass pathway activations (74,75). To improved understand the biology of ROS1 fusions and associated resistance mechanisms, researchers have employed CRISPR/Cas9 technology to model ROS1 rearrangements in vitro. Choi and Meyerson (82) designed single guide RNAs (sgRNAs) targeting intron 6 of CD74 and intron 33 of ROS1, successfully inducing a CD74-ROS1 translocation via CRISPR-mediated chromosomal inversion. This model provided a reliable platform for evaluating fusion-driven signaling and drug response. Sato et al (83) engineered EZR-ROS1 fusions in HBECp53 lung adenocarcinoma cells using CRISPR/Cas9. The resulting cells exhibited hyperactivation of the MEK/ERK signaling pathway, implicating this axis in both primary and acquired resistance to ROS1-targeted therapies. Treatment with a combination of the MEK inhibitor selumetinib and crizotinib markedly suppressed cell proliferation in vitro and in vivo, offering a promising therapeutic strategy.
MET
The MET proto-oncogene, located on chromosome 7q31, encodes the MET receptor tyrosine kinase, a key component of the hepatocyte growth factor (HGF)/MET signaling axis. Upon HGF binding, MET dimerizes and undergoes autophosphorylation, activating downstream pathways such as PI3K/AKT, RAS/ERK and Wnt/β-catenin, which regulate cell proliferation, survival, motility and invasion (84–86). In NSCLC, aberrant MET signaling is observed in 5–20% of patients and may result from gene amplification, exon 14 skipping mutations, or protein overexpression (87). MET alterations serve as both primary oncogenic drivers and mechanisms of acquired resistance to EGFR-TKIs, making MET a critical therapeutic target. Several MET inhibitors, such as crizotinib, tepotinib, capmatinib and cabozantinib, have been approved or are under clinical investigation (88,89). Despite these advances, resistance to MET-targeted therapies remains a challenge. CRISPR/Cas9 technology has been key in modeling MET-driven oncogenesis and resistance mechanisms. For example, Togashi et al (90) used CRISPR/Cas9 to introduce MET exon 14 skipping mutations in 293 cells. The edited cells exhibited increased MET protein expression, enhanced phosphorylation and heightened sensitivity to crizotinib, supporting the oncogenic potential of MET exon 14 skipping (METex14) alterations and their role as therapeutic targets. In another study, Fernandes et al (91) employed CRISPR/Cas9 to generate isogenic models of METex14 in non-transformed human lung cells. These engineered cells displayed increased tumor sphere formation, motility and invasiveness in an HGF-dependent manner. When transplanted into NSG-hHGF knock-in mice, METex14 cells formed tumors, confirming their oncogenic capability in vivo. These models also proved valuable for evaluating the efficacy of MET inhibitors and understanding resistance mechanisms. Together, these findings highlight the utility of CRISPR/Cas9 in precisely modeling MET alterations in NSCLC. Such models enable mechanistic studies of MET-driven tumorigenesis and facilitate preclinical testing of targeted therapies, ultimately contributing to the development of more effective treatment strategies.
B-Raf murine sarcoma viral oncogene homolog B (BRAF)
BRAF is a serine/threonine kinase and a key effector molecule of the MAPK/ERK signaling pathway. BRAF mutations occur in ~4% of NSCLC cases (92). Of BRAF mutations, ~50% are BRAF (V600E) (93). The BRAF (V600E) mutation leads to constitutive activation of the BRAF kinase, independent of upstream RAS signaling. This mutation enhances kinase activity by stabilizing the active conformation through a salt bridge with residue K507, resulting in sustained activation of downstream MEK-ERK signaling and promoting oncogenesis (94,95). Targeted therapies such as vemurafenib and dabrafenib have demonstrated clinical efficacy in BRAF-mutant cancers, including NSCLC. However, resistance to these inhibitors frequently emerges, driven by compensatory signaling or secondary molecular alterations (96,97). For example, reactivation of EGFR signaling or loss of the BRAF (V600E) allele has been associated with treatment failure, highlighting the need for a deeper understanding of resistance mechanisms (98). Recent CRISPR/Cas9-based studies have provided valuable insights into the biology of BRAF-mutant NSCLC. Vaishnavi et al (99) used CRISPR/Cas9 to knock out RBMS3, an RNA-binding protein, in BRAF (V600E)-driven lung-like organoids. Loss of RBMS3 enhanced tumorigenicity and resulted in micro-papillary histological features. Moreover, RBMS3 deletion conferred resistance to combination therapy with dabrafenib and trametinib, mediated through activation of the Wnt/β-catenin signaling pathway. These findings underscore the role of RBMS3 as a tumor suppressor and modulator of therapeutic response in BRAF-mutant NSCLC. CRISPR-based functional genomics thus enables precise dissection of resistance pathways and identification of novel co-targetable vulnerabilities. Collectively, CRISPR/Cas9 technology has expanded our understanding of BRAF-driven lung cancer by facilitating the development of robust experimental models and uncovering mechanisms of resistance that may guide the design of future combination therapies.
Discussion
CRISPR/Cas9 gene editing technology has emerged as a transformative tool in cancer research, offering unparalleled precision, efficiency and versatility for genomic manipulation. In the context of NSCLC, it has facilitated the identification and functional characterization of genes involved in tumorigenesis, therapeutic resistance and cellular adaptation. As targeted therapies continue to evolve as frontline treatments for molecularly defined subtypes of NSCLC, CRISPR-based strategies provide essential insights into mechanisms of resistance and opportunities for the development of more effective interventions.
With the shift from traditional cytotoxic chemotherapy to molecularly targeted therapies guided by genomic profiling, the treatment of advanced NSCLC has entered a new era of precision oncology (100–102). However, despite the initial success of EGFR-TKIs, ALK inhibitors and other targeted agents, the emergence of acquired resistance remains a major therapeutic obstacle. Mechanisms such as secondary mutations, bypass pathway activation and phenotypic transformation have all been implicated (103–105). CRISPR/Cas9 offers a powerful platform to investigate these resistance pathways. High-throughput CRISPR screens, in particular, allow for systematic identification of genes that modulate drug response. For example, genome-wide loss-of-function screens can uncover genes whose deletion sensitizes or desensitizes cancer cells to specific inhibitors, providing a foundation for rational combination therapies. Additionally, CRISPR libraries targeting regulatory elements, enhancers, or non-coding RNAs expand the scope of discovery beyond protein-coding genes. Studies (106,107) have demonstrated the utility of CRISPR/Cas9 in dissecting gene dependencies associated with various oncogenic drivers. For instance, deletion of KEAP1 in multiple NSCLC cell lines reduced sensitivity to inhibitors targeting EGFR, KRAS, BRAF and ALK, suggesting its role in broad-spectrum drug resistance through modulation of oxidative stress responses (106). Similarly, knockout of MAP2K1 (MEK1) or MET exon 14 using CRISPR markedly impaired tumor cell growth and enhanced inhibitor sensitivity, further validating these genes as critical therapeutic targets (107).
Despite its promise, CRISPR/Cas9 technology faces notable challenges. Off-target effects remain a concern, as unintended DNA cleavage may introduce undesired mutations. Although high-fidelity Cas9 variants and optimized guide RNA designs have improved specificity, complete elimination of off-target activity is yet to be achieved. Moreover, double-strand DNA breaks induced by Cas9 trigger p53-mediated DNA damage responses, potentially selecting for p53-deficient clones and increasing the risk of oncogenic transformation. Delivery remains another bottleneck, with viral vectors and nanoparticles offering variable efficiency, specificity and safety profiles.
Another critical consideration is the long-term effect of somatic cell gene editing. While ex vivo applications are progressing toward clinical translation, in vivo applications must contend with challenges related to delivery, immunogenicity and unintended systemic effects. These concerns necessitate rigorous preclinical validation and regulatory oversight to ensure safety and efficacy in therapeutic settings.
In summary, CRISPR/Cas9 technology has revolutionized our ability to model, understand potentially overcome resistance to targeted therapies in NSCLC. It serves not only as a research tool for validating therapeutic targets but also as a potential therapeutic modality itself. Continued advances in editing precision, delivery platforms and functional screening approaches will be critical for fully harnessing the potential of CRISPR-based interventions in lung cancer and beyond.
Acknowledgements
Not applicable.
Funding
The present study was supported by Startup Fund for Scientific Research, Fujian Medical University (grant no. 2023QH2024), National Natural Science Foundation of China (grant no. 82203307); Fujian provincial health technology project (grant no. 2022GGA021); Fujian Provincial Natural Science Foundation of China (grant no. 2022J01241); Talent Fund Project of Fujian Medical University Union Hospital (grant no. 2021XH029) and Joint Fund for the innovation of science and Technology, Fujian province (grant no. 2023Y9204).
Availability of data and materials
Not applicable.
Authors' contributions
JD was responsible for conceptualization, investigation and writing the original draft. XG was responsible for data curation and methodology. RH was responsible for formal analysis and visualization. BZ was responsible for investigation and methodology. CC Chen was responsible for supervision and resources. ZY was responsible for funding acquisition, writing, reviewing and editing. Data authentication is not applicable. All authors reviewed and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 74:229–263. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Siegel RL, Giaquinto AN and Jemal A: Cancer statistics, 2024. CA Cancer J Clin. 74:12–49. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Cooper AJ, Kobayashi Y, Kim D, Clifford SE, Kravets S, Dahlberg SE, Chambers ES, Li J, Rangachari D, Nguyen T, et al: Identification of a RAS-activating TMEM87A-RASGRF1 fusion in an exceptional responder to sunitinib with non-small cell lung cancer. Clin Cancer Res. 26:4072–4079. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Tsuji T, Ozasa H, Aoki W, Aburaya S, Yamamoto Funazo T, Furugaki K, Yoshimura Y, Yamazoe M, Ajimizu H, Yasuda Y, et al: YAP1 mediates survival of ALK-rearranged lung cancer cells treated with alectinib via pro-apoptotic protein regulation. Nat Commun. 11:742020. View Article : Google Scholar : PubMed/NCBI | |
|
Jamroskovic J, Doimo M, Chand K, Obi I, Kumar R, Brännström K, Hedenström M, Nath Das R, Akhunzianov A, Deiana M, et al: Quinazoline ligands induce cancer cell death through selective STAT3 inhibition and G-quadruplex stabilization. J Am Chem Soc. 142:2876–2888. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Cho SW, Kim S, Kim JM and Kim JS: Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 31:230–232. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Jinek M, East A, Cheng A, Lin S, Ma E and Doudna J: RNA-programmed genome editing in human cells. Elife. 2:e004712013. View Article : Google Scholar : PubMed/NCBI | |
|
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA and Charpentier E: A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337:816–821. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Sharma G, Sharma AR, Bhattacharya M, Lee SS and Chakraborty C: CRISPR-Cas9: A preclinical and clinical perspective for the treatment of human diseases. Mol Ther. 29:571–586. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Wu X, Ma W, Mei C, Chen X, Yao Y, Liu Y, Qin X and Yuan Y: Description of CRISPR/Cas9 development and its prospect in hepatocellular carcinoma treatment. J Exp Clin Cancer Res. 39:972020. View Article : Google Scholar : PubMed/NCBI | |
|
Meng H, Nan M, Li Y, Ding Y, Yin Y and Zhang M: Application of CRISPR-Cas9 gene editing technology in basic research, diagnosis and treatment of colon cancer. Front Endocrinol (Lausanne). 14:11484122023. View Article : Google Scholar : PubMed/NCBI | |
|
Yang F, Wang H, Fan S, Qiu H, Li X, Shi G, Li Z, Luan X and Wu H: Advances in synthetic lethality in potential oncology therapeutic approaches. Curr Top Med Chem. Jan 30–2025.(Epub ahead of print). View Article : Google Scholar | |
|
Dimitri A, Herbst F and Fraietta JA: Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 21:782022. View Article : Google Scholar : PubMed/NCBI | |
|
Akram F, Haq IU, Ahmed Z, Khan H and Ali MS: CRISPR-Cas9, a promising therapeutic tool for cancer therapy: A review. Protein Pept Lett. 27:931–944. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, Choi ALM, Mitchell DC, Swaney DL, Hann B, et al: KRASG12C inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal. 12:eaaw94502019. View Article : Google Scholar : PubMed/NCBI | |
|
Midha A, Dearden S and McCormack R: EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am J Cancer Res. 5:2892–2911. 2015.PubMed/NCBI | |
|
Thai AA, Solomon BJ, Sequist LV, Gainor JF and Heist RS: Lung cancer. Lancet. 398:535–554. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Demirci Y, Zhang B and Unver T: CRISPR/Cas9: An RNA-guided highly precise synthetic tool for plant genome editing. J Cell Physiol. 233:1844–1859. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Lattanzi A, Meneghini V, Pavani G, Amor F, Ramadier S, Felix T, Antoniani C, Masson C, Alibeu O, Lee C, et al: Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol Ther. 27:137–150. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Li Y, Ma S, Sun L, Zhang T, Chang J, Lu W, Chen X, Liu Y, Wang X, Shi R, et al: Programmable single and multiplex base-editing in bombyx mori using RNA-guided cytidine deaminases. G3 (Bethesda). 8:1701–1709. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Donohoue PD, Pacesa M, Lau E, Vidal B, Irby MJ, Nyer DB, Rotstein T, Banh L, Toh MS, Gibson J, et al: Conformational control of Cas9 by CRISPR hybrid RNA-DNA guides mitigates off-target activity in T cells. Mol Cell. 81:3637–3649.e5. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Cai W and Wang M: Engineering nucleic acid chemistry for precise and controllable CRISPR/Cas9 genome editing. Sci Bull (Beijing). 64:1841–1849. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Tian X, Gu T, Patel S, Bode AM, Lee MH and Dong Z: CRISPR/Cas9-an evolving biological tool kit for cancer biology and oncology. NPJ Precis Oncol. 3:82019. View Article : Google Scholar : PubMed/NCBI | |
|
Rotow J and Bivona TG: Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 17:637–658. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Dilly J, Hoffman MT, Abbassi L, Li Z, Paradiso F, Parent BD, Hennessey CJ, Jordan AC, Morgado M, Dasgupta S, et al: Mechanisms of resistance to oncogenic KRAS inhibition in pancreatic cancer. Cancer Discov. 14:2135–2161. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Drosten M and Barbacid M: Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell. 37:543–550. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Isermann T, Sers C, Der CJ and Papke B: KRAS inhibitors: Resistance drivers and combinatorial strategies. Trends Cancer. 11:91–116. 2025. View Article : Google Scholar : PubMed/NCBI | |
|
Ou SHI, Jänne PA, Leal TA, Rybkin II, Sabari JK, Barve MA, Bazhenova L, Johnson ML, Velastegui KL, Cilliers C, et al: First-in-human phase I/IB dose-finding study of adagrasib (MRTX849) in patients with advanced KRASG12C solid tumors (KRYSTAL-1). J Clin Oncol. 40:2530–2538. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Salgia R, Pharaon R, Mambetsariev I, Nam A and Sattler M: The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep Med. 2:1001862021. View Article : Google Scholar : PubMed/NCBI | |
|
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F, et al: Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 384:2371–2381. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Gao Q, Ouyang W, Kang B, Han X, Xiong Y, Ding R, Li Y, Wang F, Huang L, Chen L, et al: Selective targeting of the oncogenic KRAS G12S mutant allele by CRISPR/Cas9 induces efficient tumor regression. Theranostics. 10:5137–5153. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Dompe N, Klijn C, Watson SA, Leng K, Port J, Cuellar T, Watanabe C, Haley B, Neve R, Evangelista M and Stokoe D: A CRISPR screen identifies MAPK7 as a target for combination with MEK inhibition in KRAS mutant NSCLC. PLoS One. 13:e01992642018. View Article : Google Scholar : PubMed/NCBI | |
|
Li K, Yang M, Liang N and Li S: Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small cell lung cancer: Perplexity and solution (review): Perplexity and solution. Oncol Rep. 37:1347–1358. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, et al: Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 21:2237–2246. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, et al: Erlotinib in lung cancer-molecular and clinical predictors of outcome. N Engl J Med. 353:133–144. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Shi Y, Zhang L, Liu X, Zhou C, Zhang L, Zhang S, Wang D, Li Q, Qin S, Hu C, et al: Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): A randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 14:953–961. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Yang JCH, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O'Byrne K, Feng J, et al: Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 16:141–151. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, Tsuji F, Linke R, Rosell R, Corral J, et al: Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 18:1454–1466. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, et al: Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 378:113–125. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Yang JCH, Camidge DR, Yang CT, Zhou J, Guo R, Chiu CH, Chang GC, Shiah HS, Chen Y, Wang CC, et al: Safety, efficacy, and pharmacokinetics of almonertinib (HS-10296) in pretreated patients with EGFR-mutated advanced NSCLC: A multicenter, open-label, phase 1 trial. J Thorac Oncol. 15:1907–1918. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Shi Y, Hu X, Zhang S, Lv D, Wu L, Yu Q, Zhang Y, Liu L, Wang X, Cheng Y, et al: Efficacy, safety, and genetic analysis of furmonertinib (AST2818) in patients with EGFR T790M mutated non-small-cell lung cancer: A phase 2b, multicentre, single-arm, open-label study. Lancet Respir Med. 9:829–839. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Chmielecki J, Gray JE, Cheng Y, Ohe Y, Imamura F, Cho BC, Lin MC, Majem M, Shah R, Rukazenkov Y, et al: Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer. Nat Commun. 14:10702023. View Article : Google Scholar : PubMed/NCBI | |
|
Wang ZF, Ren SX, Li W and Gao GH: Frequency of the acquired resistant mutation T790 M in non-small cell lung cancer patients with active exon 19Del and exon 21 L858R: A systematic review and meta-analysis. BMC Cancer. 18:1482018. View Article : Google Scholar : PubMed/NCBI | |
|
Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, Kris MG, Pao W, Miller VA and Ladanyi M: Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 17:1169–1180. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Kuiper JL, Heideman DAM, Thunnissen E, Paul MA, van Wijk AW, Postmus PE and Smit EF: Incidence of T790M mutation in (sequential) rebiopsies in EGFR-mutated NSCLC-patients. Lung Cancer. 85:19–24. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ: Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MRV, Ward RA, Mellor MJ, et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, et al: Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Ichihara E and Lovly CM: Shades of T790M: Intratumor heterogeneity in EGFR-mutant lung cancer. Cancer Discov. 5:694–696. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, Feeney N, Sholl LM, Dahlberg SE, Redig AJ, et al: Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 4:1527–1534. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Park HR, Kim TM, Lee Y, Kim S, Park S, Ju YS, Kim M, Keam B, Jeon YK, Kim DW and Heo DS: Acquired resistance to third-generation EGFR tyrosine kinase inhibitors in patients with de novo EGFRT790M-mutant NSCLC. J Thorac Oncol. 16:1859–1871. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Vad-Nielsen J, Staunstrup NH, Kjeldsen ML, Dybdal N, Flandin G, De Stradis C, Daugaard TF, Vilsbøll-Larsen T, Maansson CT, Doktor TK, et al: Genome-wide epigenetic and mRNA-expression profiling followed by CRISPR/Cas9-mediated gene-disruptions corroborate the MIR141/MIR200C-ZEB1/ZEB2-FGFR1 axis in acquired EMT-associated EGFR TKI-resistance in NSCLC cells. Transl Lung Cancer Res. 12:42–65. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Zeng H, Castillo-Cabrera J, Manser M, Lu B, Yang Z, Strande V, Begue D, Zamponi R, Qiu S, Sigoillot F, et al: Genome-wide CRISPR screening reveals genetic modifiers of mutant EGFR dependence in human NSCLC. Elife. 8:e502232019. View Article : Google Scholar : PubMed/NCBI | |
|
Wang TH, Wu CC, Huang KY, Leu YL, Yang SC, Chen CL and Chen CY: Integrated omics analysis of non-small-cell lung cancer cells harboring the EGFR C797S mutation reveals the potential of AXL as a novel therapeutic target in TKI-resistant lung cancer. Cancers (Basel). 13:1112020. View Article : Google Scholar : PubMed/NCBI | |
|
Guernet A, Mungamuri SK, Cartier D, Sachidanandam R, Jayaprakash A, Adriouch S, Vezain M, Charbonnier F, Rohkin G, Coutant S, et al: CRISPR-barcoding for intratumor genetic heterogeneity modeling and functional analysis of oncogenic driver mutations. Mol Cell. 63:526–538. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Devarakonda S, Morgensztern D and Govindan R: Genomic alterations in lung adenocarcinoma. Lancet Oncol. 16:e342–e351. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang SS, Nagasaka M, Zhu VW and Ou SHI: Going beneath the tip of the iceberg. Identifying and understanding EML4-ALK variants and TP53 mutations to optimize treatment of ALK fusion positive (ALK+) NSCLC. Lung Cancer. 158:126–136. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al: Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 448:561–566. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Sasaki T, Rodig SJ, Chirieac LR and Jänne PA: The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. 46:1773–1780. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al: First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 371:2167–2177. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, et al: Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 13:1011–1019. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Riely GJ, Wood DE, Ettinger DS, Aisner DL, Akerley W, Bauman JR, Bharat A, Bruno DS, Chang JY, Chirieac LR, et al: Non-small cell lung cancer, version 4.2024, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 22:249–274. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Gadgeel SM, Shaw AT, Govindan R, Gandhi L, Socinski MA, Camidge DR, De Petris L, Kim DW, Chiappori A, Moro-Sibilot DL, et al: Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small-cell lung cancer. J Clin Oncol. 34:4079–4085. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Soria JC, Tan DSW, Chiari R, Wu YL, Paz-Ares L, Wolf J, Geater SL, Orlov S, Cortinovis D, Yu CJ, et al: First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet. 389:917–929. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, Ou SI, Pérol M, Dziadziuszko R, Rosell R, et al: Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 377:829–838. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Camidge DR, Kim HR, Ahn MJ, Yang JC, Han JY, Lee JS, Hochmair MJ, Li JY, Chang GC, Lee KH, et al: Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N Engl J Med. 379:2027–2039. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Shaw AT, Bauer TM, de Marinis F, Felip E, Goto Y, Liu G, Mazieres J, Kim DW, Mok T, Polli A, et al: First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N Engl J Med. 383:2018–2029. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Sullivan I and Planchard D: ALK inhibitors in non-small cell lung cancer: The latest evidence and developments. Ther Adv Med Oncol. 8:32–47. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Ma D, Zhang Y, Xing P, Hao X, Wang M, Wang Y, Shan L, Xin T, Liang H, Du Y, et al: Clinical features and outcomes of ALK rearranged non-small cell lung cancer with primary resistance to crizotinib. Thorac Cancer. 10:1213–1219. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Matikas A, Kentepozidis N, Georgoulias V and Kotsakis A: Management of resistance to crizotinib in anaplastic lymphoma kinase-positive non-small-cell lung cancer. Clin Lung Cancer. 17:474–482. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Spaans JN and Goss GD: Trials to overcome drug resistance to EGFR and ALK targeted therapies-past, present, and future. Front Oncol. 4:2332014. View Article : Google Scholar : PubMed/NCBI | |
|
Kong X, Pan P, Sun H, Xia H, Wang X, Li Y and Hou T: Drug discovery targeting anaplastic lymphoma kinase (ALK). J Med Chem. 62:10927–10954. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E, et al: In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 516:423–427. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Drilon A, Somwar R, Wagner JP, Vellore NA, Eide CA, Zabriskie MS, Arcila ME, Hechtman JF, Wang L, Smith RS, et al: A novel crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin Cancer Res. 22:2351–2358. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, Friboulet L, Huang D, Falk MD, Timofeevski S, et al: Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 368:2395–2401. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Shaw AT, Solomon BJ, Chiari R, Riely GJ, Besse B, Soo RA, Kao S, Lin CC, Bauer TM, Clancy JS, et al: Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: A multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 20:1691–1701. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Lim SM, Kim HR, Lee JS, Lee KH, Lee YG, Min YJ, Cho EK, Lee SS, Kim BS, Choi MY, et al: Open-label, multicenter, phase II study of ceritinib in patients with non-small-cell lung cancer harboring ROS1 rearrangement. J Clin Oncol. 35:2613–2618. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Dziadziuszko R, Krebs MG, De Braud F, Siena S, Drilon A, Doebele RC, Patel MR, Cho BC, Liu SV, Ahn MJ, et al: Updated integrated analysis of the efficacy and safety of entrectinib in locally advanced or metastatic ROS1 fusion-positive non-small-cell lung cancer. J Clin Oncol. 39:1253–1263. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MG, Shaw AT, de Braud F, Rolfo C, Ahn MJ, Wolf J, et al: Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 21:261–270. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Shaw AT, Ou SHI, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, et al: Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 371:1963–1971. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Huber KVM, Salah E, Radic B, Gridling M, Elkins JM, Stukalov A, Jemth AS, Göktürk C, Sanjiv K, Strömberg K, et al: Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 508:222–227. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Choi PS and Meyerson M: Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun. 5:37282014. View Article : Google Scholar : PubMed/NCBI | |
|
Sato H, Schoenfeld AJ, Siau E, Lu YC, Tai H, Suzawa K, Kubota D, Lui AJW, Qeriqi B, Mattar M, et al: MAPK pathway alterations correlate with poor survival and drive resistance to therapy in patients with lung cancers driven by ROS1 fusions. Clin Cancer Res. 26:2932–2945. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Skead G and Govender D: Gene of the month: MET. J Clin Pathol. 68:405–409. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Drilon A, Cappuzzo F, Ou SHI and Camidge DR: Targeting MET in lung cancer: Will expectations finally be MET? J Thorac Oncol. 12:15–26. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Pasquini G and Giaccone G: C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin Investig Drugs. 27:363–375. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Bubendorf L, Dafni U, Schöbel M, Finn SP, Tischler V, Sejda A, Marchetti A, Thunnissen E, Verbeken EK, Warth A, et al: Prevalence and clinical association of MET gene overexpression and amplification in patients with NSCLC: Results from the European thoracic oncology platform (ETOP) lungscape project. Lung Cancer. 111:143–149. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Fujino T, Suda K and Mitsudomi T: Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin Emerg Drugs. 25:229–249. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Mathieu LN, Larkins E, Akinboro O, Roy P, Amatya AK, Fiero MH, Mishra-Kalyani PS, Helms WS, Myers CE, Skinner AM, et al: FDA approval summary: Capmatinib and tepotinib for the treatment of metastatic NSCLC harboring MET exon 14 skipping mutations or alterations. Clin Cancer Res. 28:249–254. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Togashi Y, Mizuuchi H, Tomida S, Terashima M, Hayashi H, Nishio K and Mitsudomi T: MET gene exon 14 deletion created using the CRISPR/Cas9 system enhances cellular growth and sensitivity to a MET inhibitor. Lung Cancer. 90:590–597. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Fernandes M, Hoggard B, Jamme P, Paget S, Truong MJ, Grégoire V, Vinchent A, Descarpentries C, Morabito A, Stanislovas J, et al: MET exon 14 skipping mutation is a hepatocyte growth factor (HGF)-dependent oncogenic driver in vitro and in humanised HGF knock-in mice. Mol Oncol. 17:2257–2274. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Negrao MV, Raymond VM, Lanman RB, Robichaux JP, He J, Nilsson MB, Ng PKS, Amador BE, Roarty EB, Nagy RJ, et al: Molecular landscape of BRAF-mutant NSCLC reveals an association between clonality and driver mutations and identifies targetable non-V600 driver mutations. J Thorac Oncol. 15:1611–1623. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, Viola P, Pullara C, Mucilli F and Buttitta F: Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 29:3574–3579. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI and Rosen N: BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 28:370–383. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Degirmenci U, Wang M and Hu J: Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. 9:1982020. View Article : Google Scholar : PubMed/NCBI | |
|
Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, Souquet PJ, Smit EF, Groen HJ, Kelly RJ, et al: Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 17:642–650. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al: Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 373:726–736. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Lin L, Asthana S, Chan E, Bandyopadhyay S, Martins MM, Olivas V, Yan JJ, Pham L, Wang MM, Bollag G, et al: Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc Natl Acad Sci USA. 111:E748–E757. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Vaishnavi A, Juan J, Jacob M, Stehn C, Gardner EE, Scherzer MT, Schuman S, Van Veen JE, Murphy B, Hackett CS, et al: Transposon mutagenesis reveals RBMS3 silencing as a promoter of malignant progression of BRAFV600E-driven lung tumorigenesis. Cancer Res. 82:4261–4273. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Nishinarita N, Igawa S, Kasajima M, Kusuhara S, Harada S, Okuma Y, Sugita K, Ozawa T, Fukui T, Mitsufuji H, et al: Smoking history as a predictor of epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung cancer harboring EGFR mutations. Oncology. 95:109–115. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Fu S, Liu C, Huang Q, Fan S, Tang H, Fu X, Ai B, Liao Y and Chu Q: Estrogen receptor β1 activation accelerates resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in non-small cell lung cancer. Oncol Rep. 39:1313–1321. 2018.PubMed/NCBI | |
|
Girard N: Optimizing outcomes in EGFR mutation-positive NSCLC: Which tyrosine kinase inhibitor and when? Future Oncol. 14:1117–1132. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Lategahn J, Keul M and Rauh D: Lessons to be learned: The molecular basis of kinase-targeted therapies and drug resistance in non-small cell lung cancer. Angew Chem Int Ed Engl. 57:2307–2313. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Hsu KH, Huang YH, Tseng JS, Chen KC, Ku WH, Su KY, Chen JJW, Chen HW, Yu SL, Yang TY and Chang GC: High PD-L1 expression correlates with primary resistance to EGFR-TKIs in treatment naïve advanced EGFR-mutant lung adenocarcinoma patients. Lung Cancer. 127:37–43. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Terai H, Kitajima S, Potter DS, Matsui Y, Quiceno LG, Chen T, Kim TJ, Rusan M, Thai TC, Piccioni F, et al: ER stress signaling promotes the survival of cancer ‘persister cells’ tolerant to EGFR tyrosine kinase inhibitors. Cancer Res. 78:1044–1057. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Krall EB, Wang B, Munoz DM, Ilic N, Raghavan S, Niederst MJ, Yu K, Ruddy DA, Aguirre AJ, Kim JW, et al: KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife. 6:e189702017. View Article : Google Scholar : PubMed/NCBI | |
|
Gannon HS, Kaplan N, Tsherniak A, Vazquez F, Weir BA, Hahn WC and Meyerson M: Identification of an ‘exceptional responder’ cell line to MEK1 inhibition: Clinical implications for MEK-targeted therapy. Mol Cancer Res. 14:207–215. 2016. View Article : Google Scholar : PubMed/NCBI |










