



Zinc finger protein 695 facilitates the proliferation of colorectal cancer cells through activation of the NEK2 and PI3K/Akt/mTOR signaling pathways
- Authors:
- Published online on: July 16, 2025 https://doi.org/10.3892/or.2025.8949
- Article Number: 116
-
Copyright: © Li et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Colorectal cancer (CRC) is the third most common malignant tumor and the second leading cause of cancer-related deaths worldwide (1). In China, the incidence of CRC has been increasing due to changes in lifestyle and dietary habits (2,3). Both genetic and environmental factors contribute to the etiology of CRC, and the prognosis of patients with CRC is influenced by a variety of factors, including tumor staging, pathological type, location and size (4). A variety of factors can cause genetic and epigenetic genomic changes, and metabolic abnormality, ultimately leading to the occurrence and development of CRC (5). If diagnosed in the early stages, the 5-year survival rate is >90%, whereas the survival rate for those with distant metastases is only 10–14% (3,6); therefore, early diagnosis is critical for the prognosis of CRC. Until now, the treatment of CRC has mainly focused on traditional modalities, including surgery, radiotherapy and chemotherapy, and surgical resection remains the only possible cure for CRC (7). However, the prognosis of CRC treated with surgery is not favorable for patients with metastatic lesions. Due to inefficient treatment and the severe side effects of radiotherapy and chemotherapy, conventionally they are used as neoadjuvant and adjuvant treatments in CRC to reduce the risk of recurrence and reduce tumor size before surgery (8). New therapeutic approaches, such as immunotherapy, targeted therapy and nanomedicine, have been shown to improve treatment outcomes in patients with CRC; however, most of these options are still in clinical trials and have not yet been popularized (5). Therefore, the identification of novel biomarkers and more effective treatments with fewer side effects is urgent.
During the complex process of CRC tumorigenesis, increasing attention has been paid to the role of transcription factor dysregulation and the aberrant activation of signaling pathways in CRC initiation, invasion and metastasis (9–11). Zinc finger protein (ZNF)695 is a member of the ZNF family, which is the largest class of transcription factors, accounting for 2% of the total genes in the human body (12,13). ZNFs are widely distributed in eukaryotes, and are crucial in regulating gene expression, cell differentiation, proliferation and apoptosis. ZNFs are also involved in tumor development and progression (11,14–16). ZNF695 is located in the nucleus, and its dysregulation is relevant in the progression of tumors, including cervical squamous cell carcinoma (17), acute lymphoblastic leukemia (18) and prostate cancer (19); however, its role in CRC remains unclear.
NIMA-related kinase 2 (NEK2) is a member of the Nek family of serine/threonine kinases, which is present in the nucleus and cytoplasm, and serves essential roles in cell division and mitotic regulation throughout the cell cycle. NEK2 is considered an oncogenic factor in cancer progression and suppresses antitumor immunity through alternative mRNA splicing, decreased p53 stability and activation of certain signaling pathways (20,21). The PI3K/Akt/mTOR signaling pathway is a highly conserved signaling pathway in eukaryotic cells, consisting of a complex signaling axis with upstream regulatory factors and numerous downstream effector proteins (22). This pathway serves diverse roles in regulating various aspects of cellular physiology and pathology. PI3K/Akt/mTOR regulates various cellular processes, including cell proliferation, metabolism, apoptosis, autophagy, cell cycle progression, DNA repair and gene expression. Notably, abnormal activation of this pathway has been implicated in numerous types of cancer, including CRC (23,24). Due to its importance in tumorigenesis, there has been increasing research into this pathway (23,25,26). However, the effect of ZNF695 on the NEK2 and PI3K/Akt/mTOR signaling pathways in CRC remains unclear.
In the present study, the clinical relevance, contribution and downstream effects of ZNF695 in the development of CRC were investigated. The genes positively associated with ZNF695 in CRC tissues were analyzed using data from The Cancer Genome Atlas (TCGA). Through gain-of-function and loss-of-function experiments in CRC cells, ZNF695 was investigated as a tumor-promoting protein in CRC, highlighting its role in activating the NEK2 and PI3K/Akt/mTOR signaling pathways. The current study aimed to reveal the role of ZNF695 in CRC tumorigenesis.
Materials and methods
Analyzing the clinical relevance of ZNF695 and NEK2 using TCGA data
TCGA (http://cancergenome.nih.gov) is a public database used to analyze the clinical significance of various genes. The UALCAN (https://ualcan.path.uab.edu/analysis.html) (27), Gene Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/) (28) and Kaplan Meier plotter (https://kmplot.com/analysis/) (29) were used to assess the clinical relevance of genes in cancer based on the COAD dataset from TCGA database. The mRNA levels of ZNF695 and NEK2, and the Spearman's correlation coefficient between ZNF695 and NEK2 in patients with CRC were analyzed using the UALCAN and GEPIA databases. The association of ZNF695 with the survival of patients with CRC was analyzed using the Kaplan Meier plotter database.
Cell culture
The human normal colorectal cell line NCM460 was obtained from Hunan Fenghui Biotechnology Co., Ltd. (cat. no. CL0393). The CRC cell lines HCT-116 (cat. no. CCL-247) and HT-29 (cat. no. HTB-38) and were obtained from the American Type Culture Collection. The CRC cell line HCT-8 (cat. no. CL-0098) and 293T cells were purchased from Procell Life Science & Technology Co. Ltd. The cells were grown in RPMI 1640 complete medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin solution (Corning, Inc.). Cells were maintained in 6-well or 6-cm plates at 37°C in an incubator containing 5% CO2.
Overexpression of ZNF695
The coding sequence of the human ZNF695 gene (NM_020394.5) was cloned into a pCDH vector (Addgene, Inc.). The lentivirus was produced using the 2nd generation system. Briefly, 293T cells were transfected with pCDH-Ctrl (10 µg, without the coding sequence of ZNF695) or pCDH-ZNF695 plasmids (10 µg), and the packaging plasmids PSPAX2 (7.5 µg; Addgene, Inc.) and PDM2G (2.5 µg; Addgene, Inc.) using VigoFect (cat. no. T001; Vigorous Biotechnology Beijing Co., Ltd.) at 37°C for 48 h. Subsequently, the culture supernatants were collected, filtered, ultracentrifuged (100,000 × g at 4°C for 2 h) and the lentivirus was harvested. HT-29 cells (8×105) were infected with Ctrl and ZNF695 lentiviruses (20 µl) without detection of the titer, assisted by polybrene (8 µg/ml). After 48 h, puromycin (5 µg/ml) was used to select stable overexpressing cell lines for 1 week. After determining the overexpression efficacy by reverse transcription-quantitative PCR (RT-qPCR) and western blotting, the cells were immediately subjected to subsequent experiments.
Knockdown of ZNF695 and NEK2
Knockdown of ZNF695 and NEK2 was conducted using small interfering RNAs (siRNAs). Briefly, 8×105 HCT-8 or HT-29 cells were seeded in 6-well plates and transfected with siRNAs (30 nM) against negative control, ZNF695 or NEK2 using RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 48 h according to a previously described protocol (30). A total of 2 days after transfection the cells were subjected to other experiments, including RT-qPCR, western blotting and functional assays. siRNAs were obtained from Huzhou Hippo Biotechnology Co., Ltd. and the sequences were as follows: siCtrl, 5′-TTCTCCGAACGTGTCACGT-3′; siZNF695#1, 5′-GGATAGCTTCAATATGCAA-3′; siZNF695#2, 5′-CGATGTGAAGAATGTGGAA-3′; siNEK2, 5′-GGCTAGCTAGAATATTAAA-3′.
RT-qPCR
Total RNA was isolated from cells using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.), following the manufacturer's instruction. The RNA was subjected to RT using ReverTra Ace™ qPCR RT Master Mix with gDNA Remover kit (Toyobo Co., Ltd.), according to the manufacturer's protocol. Firstly, 4X DN Master Mix with gDNA Remover was added to the RNA samples to remove gDNA at 37°C for 5 min. Subsequently, 5X RT M aster Mix II was added and RT was conducted as follows: 37°C, 15 min; 50°C, 5 min; 98°C, 5 min; and 4°C, hold. Subsequently, qPCR was performed using SYBR master mixture (cat. no. QPK-201; Toyobo Co., Ltd.) on a Bio-Rad system (Bio-Rad Laboratories, Inc.). The qPCR procedure was performed as follows: One cycle at 37°C for 30 sec; followed by 40 cycles at 94°C for 5 sec and 60°C for 30 sec; and a final dissociation stage of one cycle at 94°C for 15 sec, 60°C for 30 sec and 94°C for 15 sec. The primer sequences used for qPCR were: ZNF695, forward 5′-ATCTCCCTTGGTGAGGATAGC-3′, reverse 5′-GACAAAACTGAGTGTTTGGCTG-3′; NEK2, forward 5′-TGCTTCGTGAACTGAAACATCC-3′, reverse 5′-CCAGAGTCAACTGAGTCATCACT-3′; β-actin, forward 5′-CATGTACGTTGCTATCCAGGC-3′ and reverse 5′-CTCCTTAATGTCACGCACGAT-3′. The relative expression levels of the indicated gene were calculated using the 2-∆∆Cq method (31).
Western blotting
Total protein was isolated from the cells using RIPA buffer (Beyotime Institute of Biotechnology) and the concentration of total proteins was detected using a BCA kit (Thermo Fisher Scientific, Inc.). Equal amounts (30–50 µg) of total proteins in loading buffer were then separated by SDS-PAGE on 10 or 12% gels and transferred onto PVDF membranes. Subsequently, the membranes were incubated with 5% skimmed milk for 1 h at room temperature, washed with PBS-0.1% Tween 20 (PBST) three times, and incubated with primary antibodies at 2–8°C overnight. After further washing with PBST three times, the membranes were incubated with secondary antibodies for 2 h at room temperature. Protein abundance was detected using the ECL-Plus kit (Amersham; Cytiva). The following primary antibodies were used: ZNF695 (cat. no. 25556-1-AP; 1:800; Proteintech Group, Inc.), NEK2 (cat. no. 24171-1-AP; 1:800; Proteintech Group Inc.), Akt (cat. no. 9272; 1:1,000; Cell Signaling Technology, Inc.), phosphorylated (p)-Akt (cat. no. 4060; 1:1,000; Cell Signaling Technology, Inc.), ribosomal protein S6 (S6; cat. no. 2217; 1:1,000; Cell Signaling Technology, Inc.), p-S6 (cat. no. 2211; 1:1,000; Cell Signaling Technology, Inc.), p21 (cat. no. 2947; 1:1,000; Cell Signaling Technology, Inc.), Bcl-2 (cat. no. 3498; 1:1,000; Cell Signaling Technology, Inc.), β-actin (cat. no. 66009-1-Ig; 1:5,000; Proteintech Group Inc.) and GAPDH (cat. no. 60004-1-Ig; 1:5,000; Proteintech Group Inc.). The mouse anti-rabbit IgG-HRP secondary antibody (cat. no. sc-2357; 1:10,000) was purchased from Santa Cruz Biotechnology, Inc. and the HRP-conjugated goat anti-mouse IgG (H+L) secondary antibody (cat. no. SA00001-1; 1:10,000) were was from Proteintech Group, Inc.
Cell viability and cytotoxicity assay
Cell viability and proliferation was assessed by detecting the relative viability of CRC cells using the Cell Counting Kit-8 (CCK8; cat. no. C0039; Beyotime Institute of Biotechnology) kit. Briefly, 3,000-4,000 HCT-8 or HT-29 cells were seeded in triplicate into 96-well plates, which contained 200 µl complete culture medium. The detection and calculation methods were carried out as previously described (30). To detect the cytotoxicity of rapamycin (cat. no. S1039; Selleck Chemicals), 3,000-4,000 HCT-8 or HT-29 cells were seeded in triplicate into 96-well plates, supplied with 200 µl complete culture medium. After 6–8 h, the cells adhered on the plates and cell culture medium was removed. Subsequently, 200 µl complete culture medium containing different concentrations of rapamycin (0, 0.1, 0.5, 2 and 5 nM) was added into each well. After incubating the cells at 37°C for 48 h, cell viability was detected using the CCK8 kit. The growth inhibition rate of 0.1/0.5/2/5 nM rapamycin was calculated as follows: Growth inhibition rate (%)=(OD450 value of 0 nM rapamycin-OD450 value of 0.1/0.5/2/5 nM rapamycin)/OD450 value of 0 nM rapamycin ×100.
Colony formation assay
After ZNF695 knockdown, a total of 1,500 siCtrl, siZNF695#1 and siZNF695#2 HCT-8 cells were seeded into 6-well plates, and were cultured in complete culture medium for 7 days. After ZNF695 overexpression with or without NEK2 knockdown, a total of 1,000 Ctrl, ZNF695-overexpressing HT-29 cells and ZNF695 + siNEK2 cells were seeded into 6-well plates, and were cultured in complete culture medium for 9 days. Subsequently, the culture medium was removed and the plates were washed three times with PBS. The colonies were then fixed with methanol for 30 min and stained with 0.1% crystal violet for 20 min at room temperature. Images of the colonies were collected using a camera (Canon EOS R100; Canon, Inc.).
Analysis of apoptosis and cell cycle using flow cytometry
A total of 1×106 HCT-8 cells transfected with siRNAs (siCtrl, siZNF695#1 and siZNF695#2) and HT-29 cells transfected with overexpression vectors (Ctrl and ZNF695) were seeded into 6-well plates. For apoptosis analysis, the cells were trypsinized in EDTA-free trypsin, and were stained with PI and Annexin V-FITC (cat. no. 40302ES60; Shanghai Yeasen Biotechnology Co., Ltd.) for 10 min at room temperature For cell cycle analysis, the cells were trypsinized and harvested in 70% iced alcohol. After 16–24 h, the cells were stained with PI and RNaseA (cat. no. 40301ES60; Shanghai Yeasen Biotechnology Co., Ltd.) for 30 min at room temperature. Apoptosis and cell cycle analyses were performed on a flow cytometry system (model no. A00-1-1102; Beckman Coulter, Inc.) at an excitation wavelength of 488 nm, and were analyzed by CytExpert software (Beckman Coulter, Inc.).
Chromatin immunoprecipitation (ChIP)-qPCR assay
The coding sequence of ZNF695 and Flag were synthesized and cloned into pCDNA3.1 vectors (Addgene, Inc.). Subsequently, 1×107 HT-29 cells in a 10-cm plate were transfected with 15 µg ZNF695-Flag-pCDNA3.1 plasmids using VigoFect at 37°C for 72 h. After 2 days, the cells were collected and subjected to ChIP-qPCR experiments using the SimpleChIP® Plus Enzymatic Chromatin IP Kit (cat. no. 9005; Cell Signaling Technology, Inc.), according to the manufacturer's instructions. The cells were divided into the ChIP-IgG group and the ChIP-Flag group; the IgG group served as the internal control. ChIP-IgG (cat. no. 2729; Cell Signaling Technology, Inc.) or ChIP-Flag (cat. no. 14793; Cell Signaling Technology, Inc.) was used during immunoprecipitation. qPCR was performed as aforementioned to quantify the amount of DNA in each group, and the qPCR primer sequences for the promoter of the NEK2 gene were: Forward, 5′-CCACTTGCAGTGCCTTACATAA-3′ and reverse, 5′-GGAATTGCCAACCAGGAGAA-3′. The qPCR procedure was performed as follows: One cycle at 37°C for 30 sec; followed by 40 cycles at 94°C for 5 sec and 60°C for 30 sec; and a final dissociation stage of one cycle at 94°C for 15 sec, 60°C for 30 sec and 94°C for 15 sec.
Measurement of caspase 3/7 activity
Caspase-Glo reagent (Promega Corporation) was used to detect the activity of caspase 3/7, according to the manufacturers' instructions. The method was carried out as previously described (30).
Dual luciferase reporter assay
The promoter sequence (−2,000-0 bp) of the NEK2 gene was cloned into the pGL3 basic vector (Promega Corporation). The Renilla luciferase vector, pCMV-RL-TK (Promega Corporation) was applied to determine the transfection efficiency. For ZNF695 knockdown, HCT-8 cells were co-transfected with siRNAs (siCtrl, siZNF695#1 and siZNF695#2) and the luciferase vectors. For ZNF695 overexpression, HT-29 cells were co-transfected with overexpression vectors (Ctrl and ZNF695) and the luciferase vectors (pGL3 vectors with NEK2 promoter and pCMV-RL-TK vectors) using Lipofectamine® 3000 (cat. no. L3000015; Invitrogen; Thermo Fisher Scientific, Inc.). Subsequently, luciferase activity was detected using an the Dual-Luciferase® Reporter Assay System (cat. no. E1910; Promega Corporation). The relative luciferase activity was normalized to Renilla luciferase.
Statistical analysis
GraphPad Prism (version 8.0; Dotmatics) was used to analyze the data (mean ± standard error) and to determine the statistical differences. Adobe Illustrator 2021 (Adobe Systems, Inc.) was used to generate the figures. Unpaired Student's t-test was applied to determine the difference between two groups. One-way ANOVA, followed by Tukey's post hoc test, was applied to compare the differences among groups. Kaplan-Meier survival curve and the log-rank test were applied to analyze overall survival. P<0.05 was considered to indicate a statistically significant difference.
Results
ZNF695 is upregulated in patients with CRC
First, the clinical significance of ZNF695 in CRC was analyzed by assessing the transcript abundance of ZNF695 in data from TCGA database. It was shown that ZNF695 mRNA expression was upregulated in the tumor tissues of most cancer types compared with non-cancer tissues (adjacent tissues from patients with cancer), including CRC (Fig. 1A and B). In addition, the cancer tissues from patients with stage 1–4 CRC exhibited higher ZNF695 expression than normal tissues (Fig. 1C). The methylation levels on the promoter of the ZNF695 gene were not significantly reduced in CRC tissues from patients of all stages compared with those in normal tissues (Fig. 1D). Notably, the cancer tissues from patients with stage 2 CRC only exhibited significantly decreased methylation levels on the promoter of the ZNF695 gene compared with those in normal samples (Fig. 1E). Furthermore, the prognostic value of ZNF695 for patients with CRC was analyzed using the Kaplan Meier plotter database. Using the Kaplan-Meier plotter, when setting the ‘best cutoff, OS, 18 months’, the patients with high expression of ZNF695 (n=406) tended to have a shorter survival time than those with low ZNF695 expression (n=655) (Fig. 1F), but the difference was not statistically significant (P=0.065). Subsequently, experiments were conducted to assess the expression of ZNF695 in NCM460 normal colorectal cells and in CRC cells. Compared with those in NCM460 cells, the mRNA levels of ZNF695 were increased in HT-29, HCT-8 and HCT116 CRC cells (Fig. 1G). Taken together, these results indicated that ZNF695 may participate in CRC development.
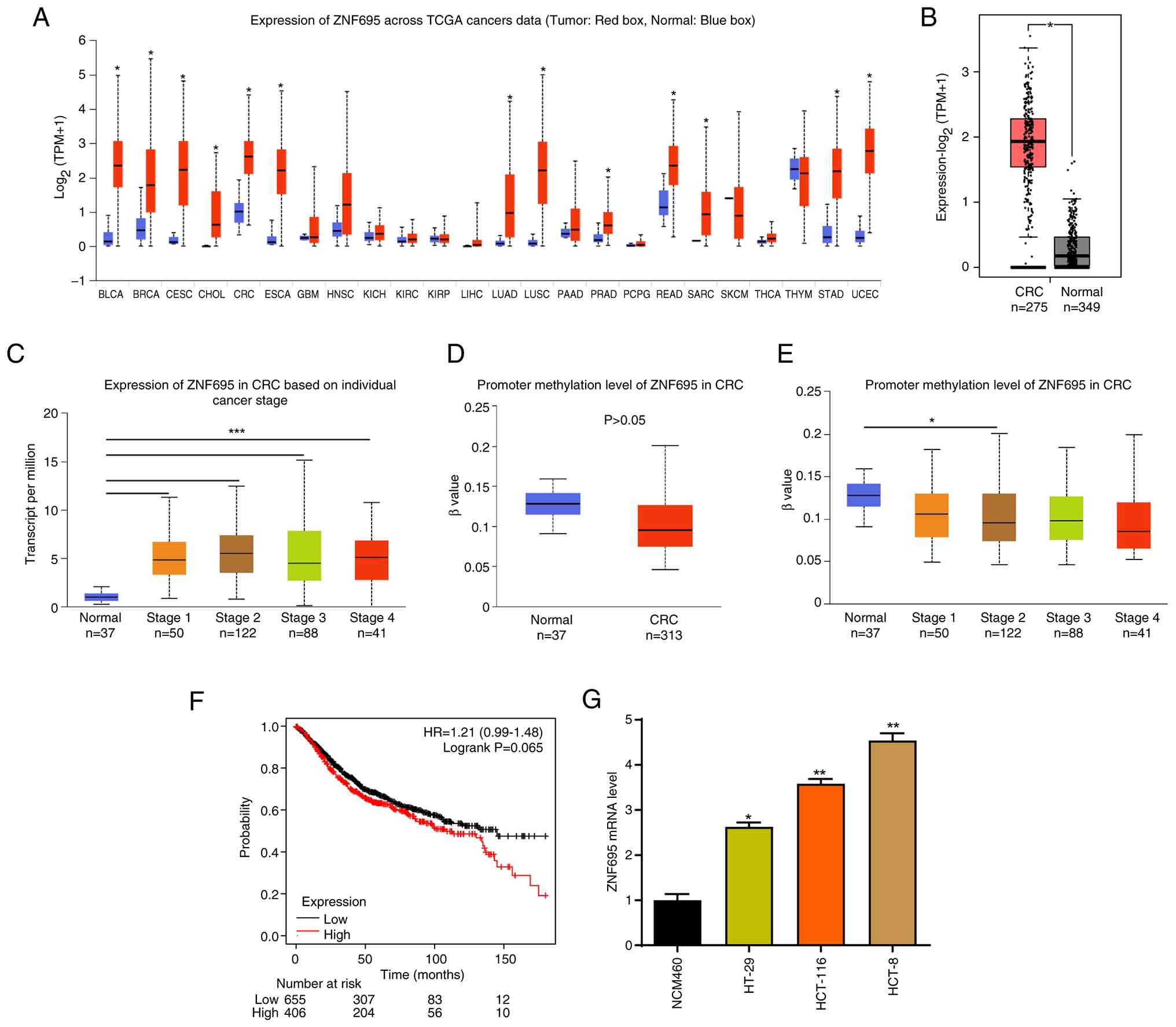
Overexpression of ZNF695 facilitates the proliferation of CRC cells
Next, whether overexpression of ZNF695 contributes to the malignant proliferation of CRC cells was investigated; to address this, HCT-8 and HT-29 cells were used. HCT-8 is a stem-like cell line, which has characteristics of fast proliferation and poor differentiation (32,33), whereas HT-29 is a moderately differentiated colonic cancer cell line (33). Using these two cell lines could demonstrate the response of CRC cells to the effect of ZNF695 expression on proliferation. ZNF695 was knocked down in HCT-8 cells by transfecting the cells with siZNF695. Notably, the mRNA and protein levels of ZNF695 were significantly reduced in siZNF695#1 and siZNF695#2 HCT-8 cells compared with those in siCtrl cells (Fig. 2A). Furthermore, knockdown of ZNF695 suppressed the proliferation of HCT-8 cells, as demonstrated by the CCK8 assay (Fig. 2B). To validate the oncogenic function of ZNF695, ZNF695 was overexpressed in HT-29 cells, and the overexpression efficacy was confirmed by RT-qPCR and western blotting (Fig. 2C). Notably, overexpression of ZNF695 promoted the proliferation of HT-29 cells (Fig. 2D). Furthermore, it was shown that ZNF695 knockdown inhibited the colony formation of HCT-8 cells, whereas the opposite results were observed in HT-29 cells with overexpression of ZNF695 (Fig. 2E and F). Thus, the in vitro results suggested that ZNF695 may be critical for the of CRC cells.
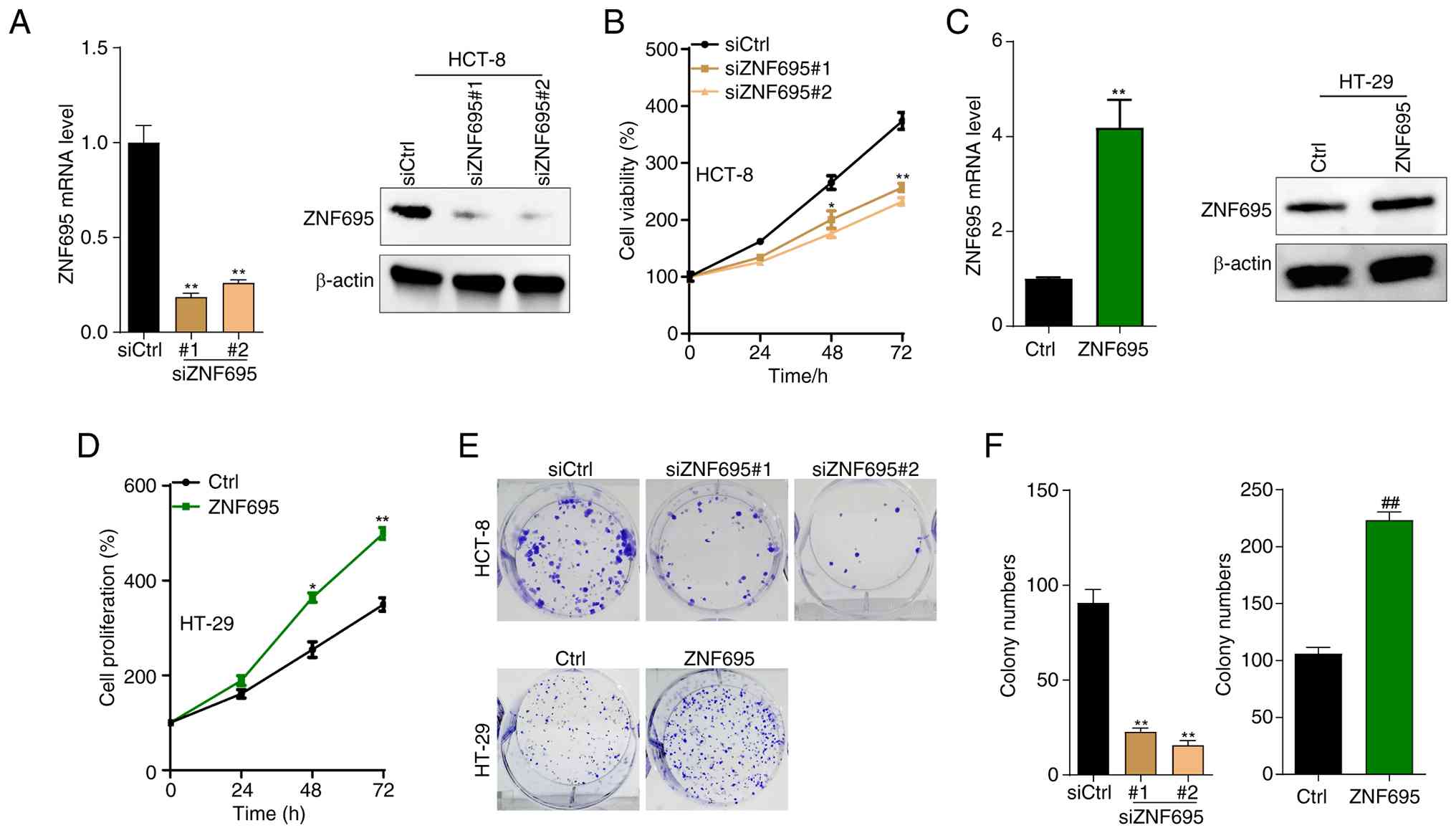
ZNF695 regulates the cell cycle and apoptosis of CRC cells
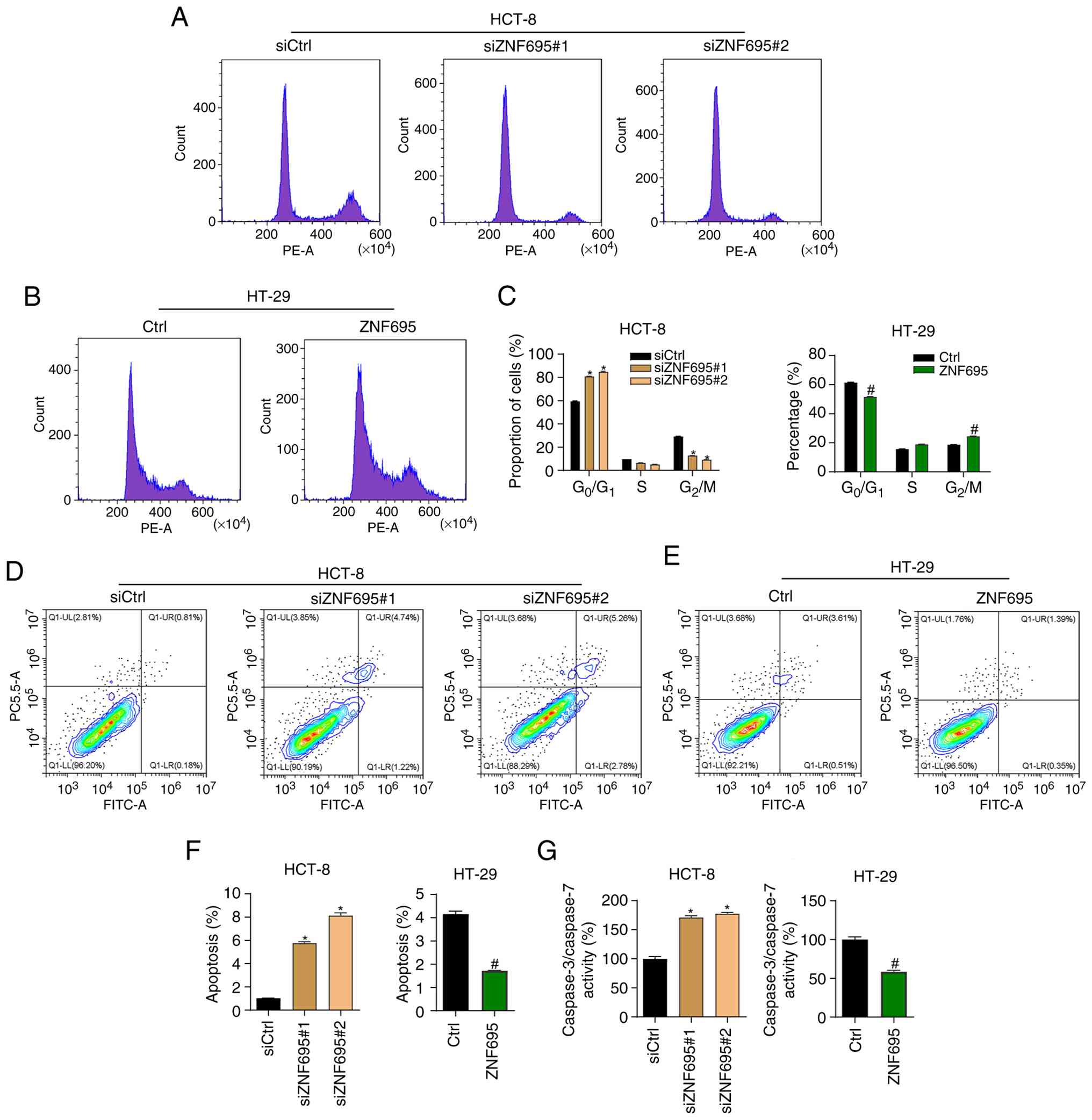
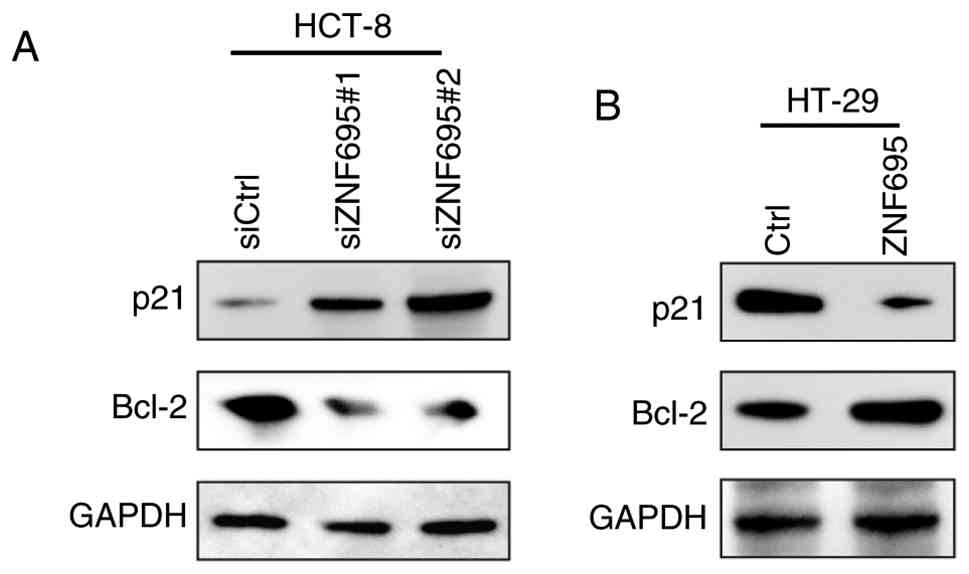
To assess whether ZNF695 regulates the cell cycle progression of CRC cells, the cells were subjected to knockdown or overexpression of ZNF695 and then underwent PI staining, followed by cell cycle analysis with flow cytometry. It was found that ZNF695 knockdown resulted in a higher percentage of HCT-8 cells in G0/G1 phase and a lower percentage in G2/M phase compared with those in the siCtrl group (Fig. 3A and C). By contrast, overexpression of ZNF695 exhibited the opposite effect on the cell cycle progression of HT-29 cells (Fig. 3B and C). Next, the cells were stained with Annexin V/PI and apoptosis was analyzed by flow cytometry. The results showed that knockdown of ZNF695 induced the apoptosis of HCT-8 cells compared with in the siCtrl group (Fig. 3D and F). Consistently, overexpression of ZNF695 suppressed the apoptosis of HT-29 cells (Fig. 3E and F). In addition, the activity of caspase 3/7 was inhibited by ZNF695 overexpression and increased by ZNF695 knockdown (Fig. 3G). Subsequently, the expression levels of the apoptosis markers p21 and Bcl-2 were assessed in cells with knockdown or overexpression of ZNF695 by western blotting. The results supported the indication that ZNF695 can suppress the apoptosis of CRC cells (Fig. 4). Collectively, these findings suggested that ZNF695 may be involved in regulating the cell cycle and apoptosis of CRC cells.
ZNF695 activates the expression of the NEK2 gene
To investigate the downstream effector of ZNF695 in CRC, the present study firstly analyzed the genes positively correlated with ZNF695 in patients with CRC. Spearman correlation results showed that C17orf53, UBE2T, NEK2, CKS1B and BIRC5 were the most significantly correlated genes with ZNF695 in patients with CRC (Fig. 5A). Previous studies have demonstrated that upregulation of NEK2 contributes to the development of various types of cancer, including CRC (34–36); therefore, the current study explored whether ZNF695 contributed to CRC progression by regulating NEK2. To address this RT-qPCR and western blotting were performed. It was shown that the abundance of NEK2 mRNA was downregulated in CRC cells with ZNF695 knockdown, whereas it was upregulated when ZNF695 was overexpressed (Fig. 5B and C). Dual luciferase reporter assay results revealed that the luciferase activity of the NEK2 gene promoter was reduced in CRC cells with ZNF695 knockdown, whereas it was increased in CRC cells overexpressing ZNF695 (Fig. 5D and E). The ChIP assay also showed that ZNF695 activated the expression of the NEK2 gene (Fig. 5F). Based on TCGA database, there was a positive correlation between ZNF695 and NEK2 in CRC samples (Fig. 5G). Furthermore, the transcript levels of NEK2 were enhanced in CRC tissues compared with those in non-cancer tissues (Fig. 5H). These findings indicated that ZNF695 may positively regulate the expression of NEK2 in CRC cells and in patients with CRC.
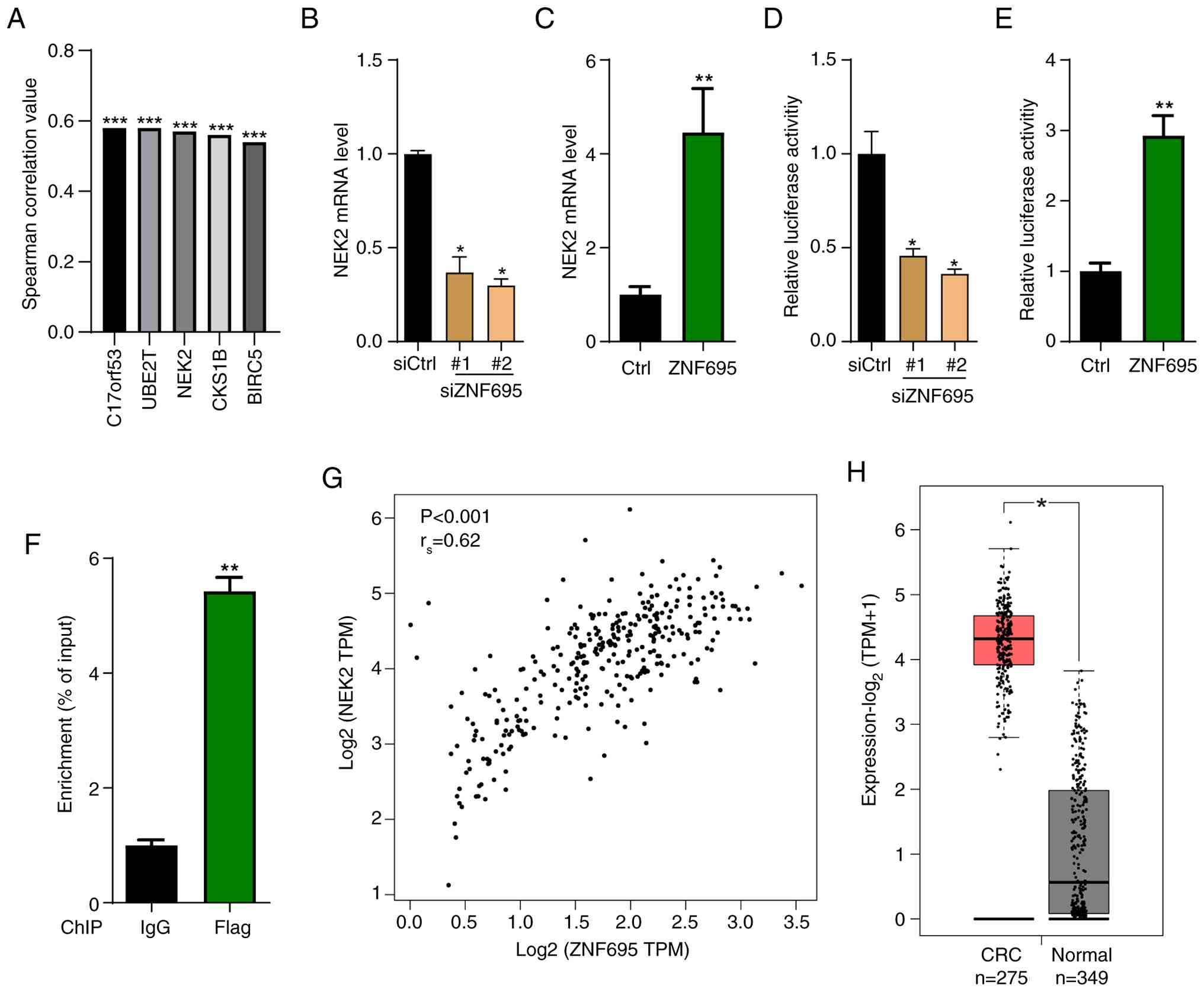
ZNF695 activates the Akt/mTOR signaling pathway in CRC cells
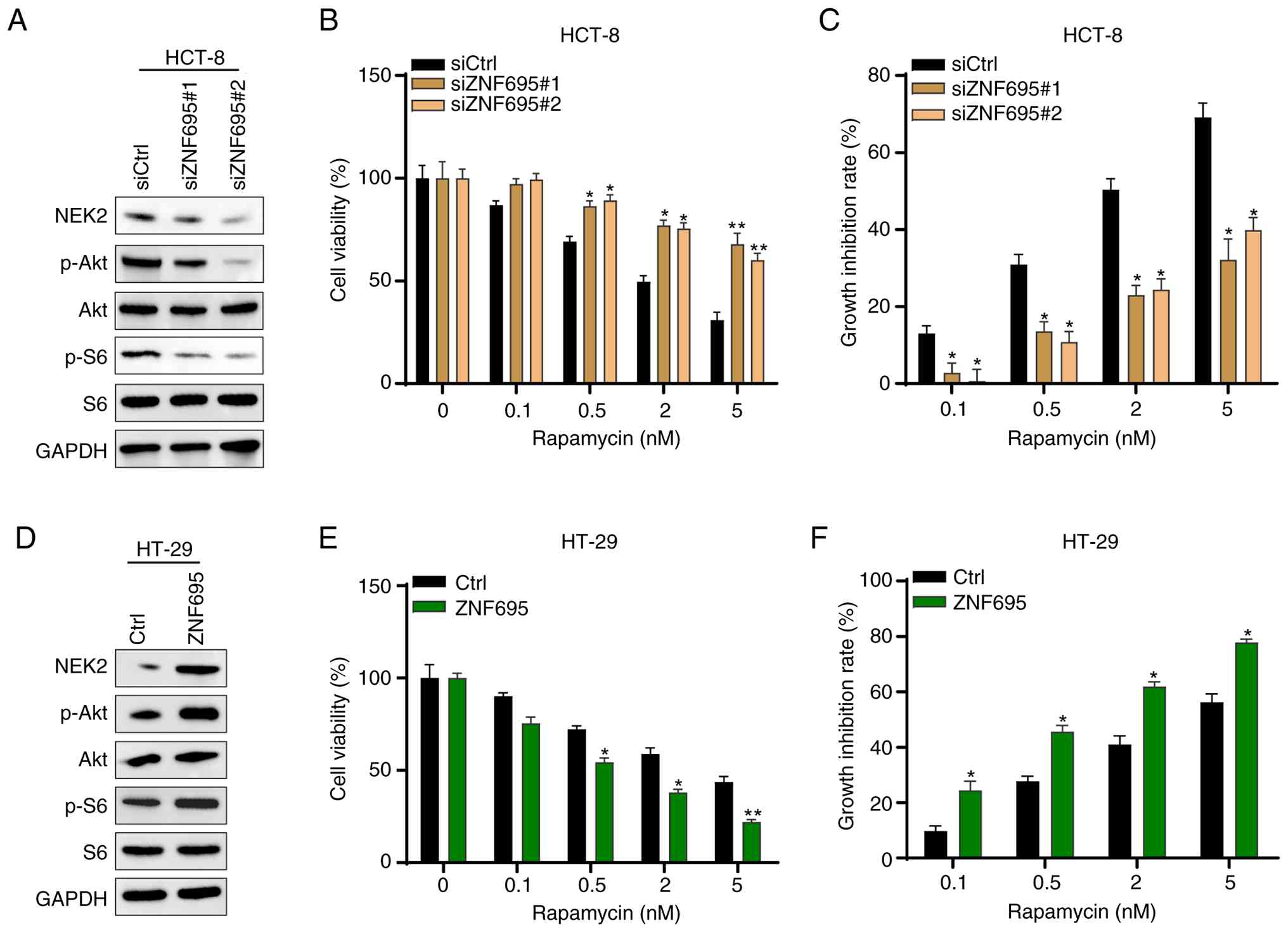
NEK2 is a kinase and thus has the potential to enhance the phosphorylation of certain proteins. The Akt/mTOR signaling pathway is hyper-activated in various types of cancer, including CRC (37). Therefore, whether ZNF695 regulates the activity of the Akt/mTOR signaling pathway in CRC cells was investigated. Western blotting showed that ZNF695 knockdown reduced the phosphorylation levels, but not the protein levels, of Akt and S6 in HCT-8 cells (Fig. 6A), whereas the opposite results were observed in HT-29 cells with overexpression of ZNF695 (Fig. 6D). Subsequently, the cells were treated with different concentrations of the mTOR inhibitor, rapamycin. It was shown that different doses of rapamycin exhibited reduced cytotoxic effects and increased growth inhibition in cells with ZNF695 knockdown as compared with siCtrl HCT-8 cells (Fig. 6B and C). In addition, HT-29 cells with ZNF695 overexpression exhibited higher sensitivity and growth inhibition comparing with Ctrl HT-29 cells in response to treatment with rapamycin at different doses (Fig. 6E and F). These results indicated that ZNF695 may activate the Akt/mTOR signaling pathway and the expression of ZNF695 dictates the sensitivity of CRC cells to rapamycin treatment.
ZNF695 facilitates the proliferation of CRC cells through upregulation of NEK2
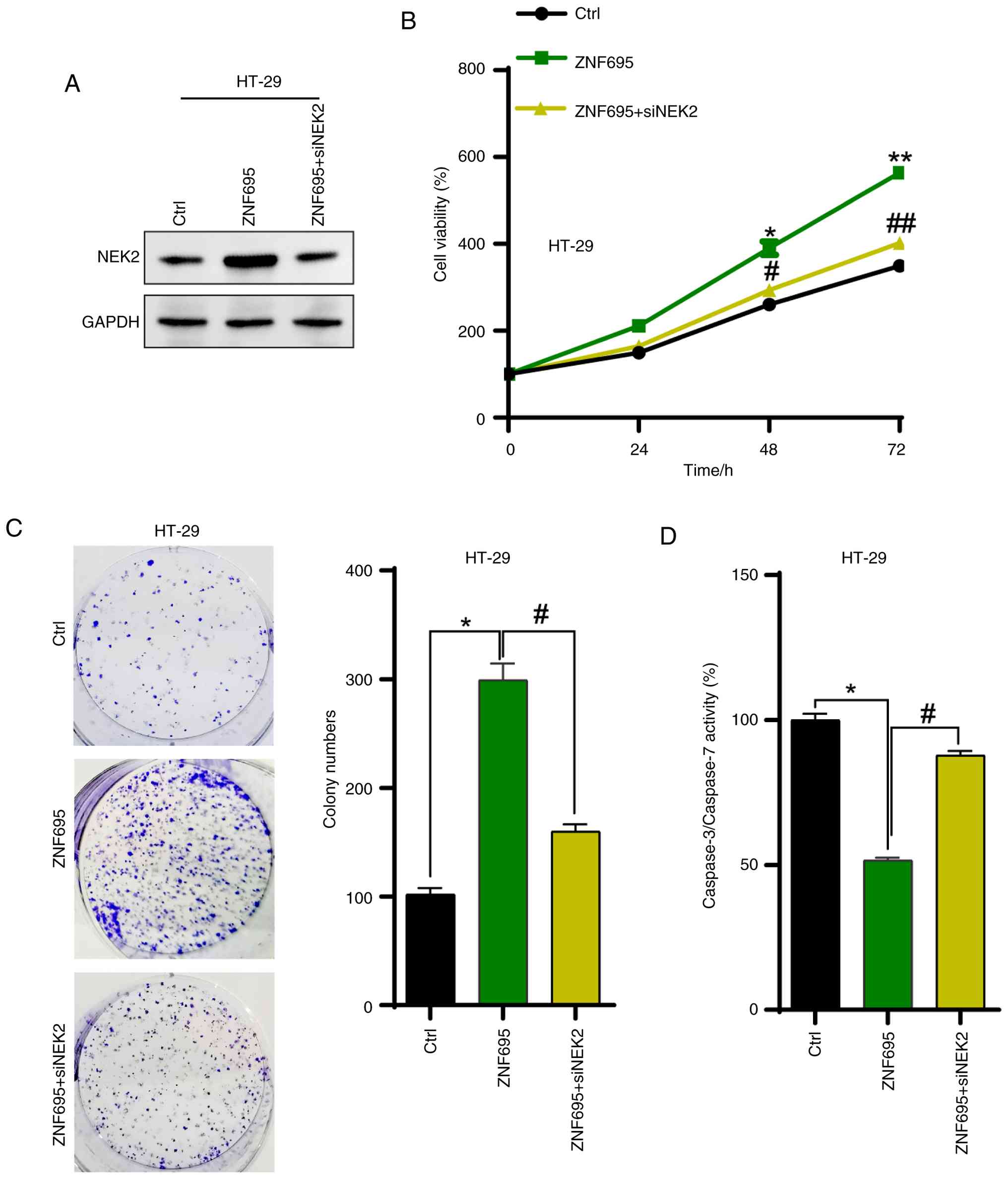
Finally, whether upregulation of NEK2 supports the proliferation of CRC cells driven by ZNF695 was investigated by knocking down NEK2 in CRC cells. The western blotting results showed that NEK2 was efficiently silenced by siRNAs in HT-29 cells (Fig. S1). NEK2 was knocked down in CRC cells with overexpression of ZNF695. Western blotting results showed that NEK2 was efficiently silenced by siRNA (Fig. 7A). CCK8 and colony formation assay results revealed that the accelerated proliferation of HT-29 cells was inhibited by knockdown of NEK2 (Fig. 7B and C). Furthermore, the activity of caspase 3/7 was enhanced by silencing NEK2 in ZNF695-overexpressing cells (Fig. 7D). These results indicated that ZNF695 may contribute to CRC cell proliferation via the upregulation of NEK2.
Discussion
The present study provided information on the key role of ZNF695 in the pathogenesis of CRC. The results demonstrated that ZNF695 was significantly upregulated in CRC tissues and cell lines compared with in their non-cancerous counterparts. Functional assays revealed that ZNF695 promoted CRC cell proliferation and survival, primarily by modulating the expression of NEK2 and activating the PI3K/Akt/mTOR signaling pathway. These findings provide evidence suggesting that ZNF695 may be a tumor-promoting transcription factor in CRC. To the best of our knowledge, the present study is the first to report the effect of ZNF695 on the proliferation of CRC cells. The findings align with and expand upon previous research highlighting the role of ZNFs in cancer progression (11,38). ZNFs, including ZNF695, are known to regulate transcriptional activity and are frequently dysregulated in various types of cancer. For example, ZNF695 has been implicated in the pathogenesis of acute lymphoblastic leukemia (39), prostate cancer (19), breast cancer (40) and other types of cancer (17), and it supports tumor growth and immune evasion. The results of the current study provide novel insights into the role of ZNF695 in integrating upstream transcriptional regulation with downstream signaling cascades, thereby contributing to CRC progression.
In the present study, the oncogenic role of ZNF695 was suggested to be mediated by its ability to upregulate NEK2 at both transcriptional and protein levels. Furthermore, ZNF695 enhanced NEK2 promoter activity, establishing a direct regulatory relationship. Furthermore, silencing NEK2 in ZNF695-overexpressing CRC cells abrogated the enhanced proliferative and survival capabilities of these cells, highlighting the functional dependency of ZNF695 on NEK2. NEK2 has an important role during normal cell mitosis by regulating centrosome separation, centrosome maturation, microtubule organization, chromosome congression and chromosome separation (20). The expression of NEK2 is regulated by transcription factor LIN9 (41), orthodenticle homeobox 2, c-MYC (42) and E2F8 (43), and is also mediated by ubiquitination (44), DNA methylation (45). Notably, circular (circ)RNA/long noncoding (lnc)RNA-microRNA (miR) axes, such as circRNA TNPO3-miR-1299, circRNA PITX1-miR-329-3p and lncRNA SNHG1-miR-195, also participate in the regulation of NEK2 expression (20,46,47). NEK2 has been established as a critical driver of oncogenesis in multiple malignancies, including CRC, promoting cell division and suppressing apoptotic pathways (20,48). The possible mechanisms underlying the inhibition of apoptosis by upregulation of NEK2 in CRC are related to several pathways, including inhibition of pro-apoptotic gene expression, such as serine/arginine-rich splicing factor 1 (49), inhibition of p53 signaling and activation of Akt signaling. Both p53 and Akt signaling are important regulators of cell apoptosis (21,50). Neal et al (51) reported that overexpression of NEK2 is associated with β-catenin relocalization from the plasma membranes to the cytoplasm and nucleus, and shortened cancer-specific survival. Ko et al (36) demonstrated that ribosomal protein L17 promotes CRC proliferation and stemness through NEK2/β-catenin signaling pathways. In addition, NEK2 phosphorylates RhoGDI1 to promote cell proliferation, migration and invasion through the activation of RhoA and Rac1 in CRC cells (52). Takahashi et al (53) also reported that upregulation of NEK2 due to miR-128 methylation is associated with poor prognosis in CRC. This previous study showed that miR-128, as a tumor suppressor, could directly target NEK2 mRNA by binding to the 3′-UTR of NEK2 transcripts and inhibiting its expression, resulting in G2-phase cell cycle arrest and the inhibition of cell proliferation in CRC. By contrast, the tumor-suppressor function of miR-128 could be silenced by DNA methylation in the promoter region of miR-128 in CRC cell lines. By elucidating the functional relationship between ZNF695 and NEK2 in CRC, the current study provides new understanding of their combined contributions to tumorigenesis.
In the present study, ZNF695 was shown to modulate the PI3K/Akt/mTOR pathway, as evidenced by increased phosphorylation of Akt and S6 proteins in cells overexpressing ZNF695, although the effect of ZNF695 on PI3K was not assessed. The PI3K/Akt/mTOR pathway orchestrates numerous cellular processes, including proliferation, survival and metabolism, and its dysregulation is a hallmark of numerous types of cancer (23). Both Akt and S6 proteins are involved in CRC tumorigenesis. Akt is the dominant effector of several downstream signaling proteins activated by PI3K signaling, and Akt regulates mTOR, a downstream target that promotes protein translation and proliferation in CRC. Furthermore, Akt promotes cell survival in CRC through the regulation of various downstream pro-survival targets, including NF-κB and XIAP, and inhibiting pro-apoptotic targets such as Bad (26). mTORC1 phosphorylates and activates S6, and S6 promotes protein synthesis and cellular proliferation; however, the signaling pathways downstream of S6 are currently largely unknown (54). It has been reported that S6 is involved in the biopharmaceutical drug resistance of CRC cell lines to selumetinib (55).
In the present study, CRC cells with high ZNF695 expression exhibited increased sensitivity to rapamycin, an mTOR inhibitor, suggesting a potential therapeutic avenue for targeting tumors overexpressing ZNF695. These findings highlight the mechanism by which ZNF695 integrates transcriptional regulation and signal transduction to promote CRC progression and identify possible targets for therapeutic intervention. NEK2 is involved in oncogenic signaling pathways, including PI3K/Akt/mTOR. Notably, it has been reported that NEK2 activates the PI3K/Akt/mTOR signaling pathway to promote cell proliferation and drug resistance in gastric cancer (56), myeloma (50) and hepatocellular carcinoma (57). In the current study, ZNF695 promoted CRC proliferation not only through NEK2, but also by activating the PI3K/Akt/mTOR pathway; therefore, it could be hypothesized that the existence of a ZNF695-NEK2-PI3K/Akt/mTOR axis drives CRC pathogenesis. Further studies are needed to validate this axis.
The significant upregulation of ZNF695 in CRC tissues positions ZNF695 as a promising biomarker for CRC prognosis. Patients with high ZNF695 expression could potentially benefit from personalized treatment strategies targeting ZNF695, NEK2 or Akt. Moreover, the observed sensitivity of ZNF695-overexpressing CRC cells to mTOR inhibition provides possibilities for incorporating mTOR inhibitors into treatment regimens for such patients. This approach could enhance the efficacy of conventional therapies by exploiting vulnerabilities in ZNF695-driven tumors. Additionally, NEK2, as a downstream effector of ZNF695, presents another therapeutic target. NEK2 inhibitors, currently in preclinical and early-phase trials (20), could be explored for their efficacy in CRC models characterized by high ZNF695 expression. A dual-targeting approach aimed at both ZNF695 and NEK2 could potentially yield synergistic antitumor effects and improve therapeutic outcomes in patients with CRC.
While the current study provides evidence for the oncogenic role of ZNF695 in CRC, it also has certain limitations. First, the findings are predominantly derived from in vitro experiments, which, while informative, do not fully capture the complexity of CRC biology in vivo. Future studies should employ animal models and clinical samples to validate the role of ZNF695 and its downstream effectors in tumorigenesis. Second, the molecular mechanisms by which ZNF695 regulates NEK2 expression remain incompletely understood. Investigating the involvement of co-factors, chromatin remodeling complexes and post-translational modifications of ZNF695 could provide deeper insights into its regulatory functions. Additionally, exploring the broader transcriptional network influenced by ZNF695 could reveal other potential oncogenic targets and pathways. Finally, the role of ZNF695 in therapeutic resistance and metastasis, major challenges in CRC management, remains unexplored. Addressing these aspects could enhance the understanding of the full spectrum of oncogenic activities of ZNF695 and uncover additional therapeutic vulnerabilities.
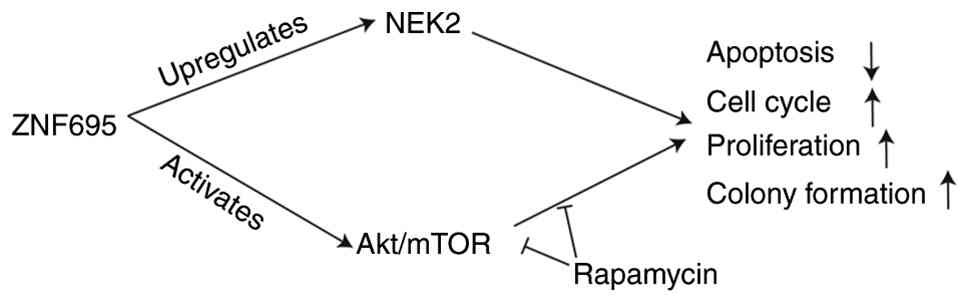
In conclusion, the current study identified ZNF695 as a novel driver of CRC progression through its regulation of NEK2 and activation of the PI3K/Akt/mTOR signaling pathway (Fig. 8). These findings highlight the potential of ZNF695 as both a biomarker for diagnosis and prognosis, as well as a therapeutic target in CRC. Targeting ZNF695, NEK2 or Akt could pave the way for innovative treatment strategies, ultimately improving outcomes for patients with CRC.
Supplementary Material
Supporting Data
Acknowledgments
Not applicable.
Funding
This study was supported by the Scientific Research Fund of Beijing Rehabilitation Hospital, Capital Medical University (grant nos. 2020-056, 2022-057 and 2023-10) and the Scientific Research Fund of Beijing Anorectal Society (grant no. 2020ABCP002).
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
QG, CK, HS and XiL designed the study. XiL, XuL, MX, MZ, JZ, YW, SH, JW, JS and XC performed the majority of the experiments. QG, CK, XiL and XuL analyzed the data. DD, YW, JW and XC provided assistance for the experiments and data analysis. QG, CK, XiL and HYS wrote the draft of the manuscript. QG revised the manuscript. QG, CK and XL confirm the authenticity of all the raw data. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.PubMed/NCBI | |
|
Xia C, Dong X, Li H, Cao M, Sun D, He S, Yang F, Yan X, Zhang S, Li N and Chen W: Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin Med J (Engl). 135:584–590. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Li N, Lu B, Luo C, Cai J, Lu M, Zhang Y, Chen H and Dai M: Incidence, mortality, survival, risk factor and screening of colorectal cancer: A comparison among China, Europe, and northern America. Cancer Lett. 522:255–268. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Housini M, Dariya B, Ahmed N, Stevens A, Fiadjoe H, Nagaraju GP and Basha R: Colorectal cancer: Genetic alterations, novel biomarkers, current therapeutic strategies and clinical trials. Gene. 892:1478572024. View Article : Google Scholar : PubMed/NCBI | |
|
Shin AE, Giancotti FG and Rustgi AK: Metastatic colorectal cancer: Mechanisms and emerging therapeutics. Trends Pharmacol Sci. 44:222–236. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Shinji S, Yamada T, Matsuda A, Sonoda H, Ohta R, Iwai T, Takeda K, Yonaga K, Masuda Y and Yoshida H: Recent advances in the treatment of colorectal cancer: A review. J Nippon Med Sch. 89:246–254. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Abedizadeh R, Majidi F, Khorasani HR, Abedi H and Sabour D: Colorectal cancer: A comprehensive review of carcinogenesis, diagnosis, and novel strategies for classified treatments. Cancer Metastasis Rev. 43:729–753. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Ionescu VA, Gheorghe G, Bacalbasa N, Chiotoroiu AL and Diaconu C: Colorectal cancer: From risk factors to oncogenesis. Medicina (Kaunas). 59:16462023. View Article : Google Scholar : PubMed/NCBI | |
|
Xu H, Liu L, Li W, Zou D, Yu J, Wang L and Wong CC: Transcription factors in colorectal cancer: Molecular mechanism and therapeutic implications. Oncogene. 40:1555–1569. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Jen J and Wang YC: Zinc finger proteins in cancer progression. J Biomed Sci. 23:532016. View Article : Google Scholar : PubMed/NCBI | |
|
Cassandri M, Smirnov A, Novelli F, Pitolli C, Agostini M, Malewicz M, Melino G and Raschellà G: Zinc-finger proteins in health and disease. Cell Death Discov. 3:170712017. View Article : Google Scholar : PubMed/NCBI | |
|
Krishna SS, Majumdar I and Grishin NV: Structural classification of zinc fingers: Survey and summary. Nucleic Acids Res. 31:532–550. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Huntley S, Baggott DM, Hamilton AT, Tran-Gyamfi M, Yang S, Kim J, Gordon L, Branscomb E and Stubbs L: A comprehensive catalog of human KRAB-associated zinc finger genes: Insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 16:669–677. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Ecco G, Imbeault M and Trono D: KRAB zinc finger proteins. Development. 144:2719–2729. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao J, Wen D, Zhang S, Jiang H and Di X: The role of zinc finger proteins in malignant tumors. FASEB J. 37:e231572023. View Article : Google Scholar : PubMed/NCBI | |
|
Ding X, Wan A, Qi X, Jiang K, Liu Z and Chen B: ZNF695, A potential prognostic biomarker, correlates with im mune infiltrates in cervical squamous cell carcinoma and endoce rvical adenocarcinoma: Bioinformatic analysis and experimental verification. Curr Gene Ther. 24:441–452. 2024. View Article : Google Scholar : PubMed/NCBI | |
|
Li C, Kuang L, Zhu B, Chen J, Wang X and Huang X: Identification of prognostic risk factors of acute lymphoblastic leukemia based on mRNA expression profiling. Neoplasma. 64:494–501. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Ke ZB, You Q, Chen JY, Sun JB, Xue YT, Zhuang RB, Zheng QS, Chen YH, Wei Y, Sun XL, et al: A radiation resistance related index for biochemical recurrence and tumor immune environment in prostate cancer patients. Comput Biol Med. 146:1057112022. View Article : Google Scholar : PubMed/NCBI | |
|
Xia J, Zhao H, Edmondson JL, Koss B and Zhan F: Role of NEK2 in tumorigenesis and tumor progression. Trends Mol Med. 31:79–93. 2025. View Article : Google Scholar : PubMed/NCBI | |
|
Choi BK, Dayaram T, Parikh N, Wilkins AD, Nagarajan M, Novikov IB, Bachman BJ, Jung SY, Haas PJ, Labrie JL, et al: Literature-based automated discovery of tumor suppressor p53 phosphorylation and inhibition by NEK2. Proc Natl Acad Sci USA. 115:10666–10671. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Ersahin T, Tuncbag N and Cetin-Atalay R: The PI3K/AKT/mTOR interactive pathway. Mol Biosyst. 11:1946–1954. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, Eng H, Nair MG, Makvandi P, Geoerger B, et al: PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 22:1382023. View Article : Google Scholar : PubMed/NCBI | |
|
Fleming-de-Moraes CD, Rocha MR, Tessmann JW, de Araujo WM and Morgado-Diaz JA: Crosstalk between PI3K/Akt and Wnt/β-catenin pathways promote colorectal cancer progression regardless of mutational status. Cancer Biol Ther. 23:1–13. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Yu L, Wei J and Liu P: Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol. 85:69–94. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Leiphrakpam PD and Are C: PI3K/Akt/mTOR signaling pathway as a target for colorectal cancer treatment. Int J Mol Sci. 25:31782024. View Article : Google Scholar : PubMed/NCBI | |
|
Chandrashekar DS, Karthikeyan SK, Korla PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne U, et al: UALCAN: An update to the integrated cancer data analysis platform. Neoplasia. 25:18–27. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Tang Z, Li C, Kang B, Gao G, Li C and Zhang Z: GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45((W1)): W98–W102. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Győrffy B: Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation (Camb). 5:1006252024.PubMed/NCBI | |
|
Zhang J, Wang Y, Hou S, Chi X, Ding D, Xue M, Zhang M, Wang J, Shuai J, Sun H, et al: Overexpression of ZNF169 promotes the growth and proliferation of colorectal cancer cells via the upregulation of ANKZF1. Oncol Rep. 51:822024. View Article : Google Scholar : PubMed/NCBI | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Tompkins WA, Watrach AM, Schmale JD, Schultz RM and Harris JA: Cultural and antigenic properties of newly established cell strains derived from adenocarcinomas of the human colon and rectum. J Natl Cancer Inst. 52:1101–1110. 1974. View Article : Google Scholar : PubMed/NCBI | |
|
Bhattacharya A, Tóth K, Sen A, Seshadri M, Cao S, Durrani FA, Faber E, Repasky EA and Rustum YM: Inhibition of colon cancer growth by methylselenocysteine-induced angiogenic chemomodulation is influenced by histologic characteristics of the tumor. Clin Colorectal Cancer. 8:155–162. 2009.PubMed/NCBI | |
|
Huang X, Zhang G, Tang T, Gao X and Liang T: One shoot, three birds: Targeting NEK2 orchestrates chemoradiotherapy, targeted therapy, and immunotherapy in cancer treatment. Biochim Biophys Acta Rev Cancer. 1877:1886962022. View Article : Google Scholar : PubMed/NCBI | |
|
Cui F, Chen Y, Wu X and Zhao W: Mesenchymal stem cell-derived exosomes carrying miR-486-5p inhibit glycolysis and cell stemness in colorectal cancer by targeting NEK2. BMC Cancer. 24:13562024. View Article : Google Scholar : PubMed/NCBI | |
|
Ko MJ, Seo YR, Seo D, Park SY, Seo JH, Jeon EH, Kim SW, Park KU, Koo DB, Kim S, et al: RPL17 promotes colorectal cancer proliferation and stemness through ERK and NEK2/β-catenin signaling pathways. J Cancer. 13:2570–2583. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Stefani C, Miricescu D, Stanescu-Spinu II, Nica RI, Greabu M, Totan AR and Jinga M: Growth factors, PI3K/AKT/mTOR and MAPK signaling pathways in colorectal cancer pathogenesis: Where are we now? Int J Mol Sci. 22:102602021. View Article : Google Scholar : PubMed/NCBI | |
|
Ye Q, Liu J and Xie K: Zinc finger proteins and regulation of the hallmarks of cancer. Histol Histopathol. 34:1097–1109. 2019.PubMed/NCBI | |
|
De la Rosa R, Villegas-Ruíz V, Caballero-Palacios MC, Pérez-López EI, Murata C, Zapata-Tarres M, Cárdenas-Cardos R, Paredes-Aguilera R, Rivera-Luna R and Juárez-Méndez S: Expression of ZNF695 transcript variants in childhood B-cell acute lymphoblastic leukemia. Genes (Basel). 10:7162019. View Article : Google Scholar | |
|
Li R, Campos J and Iida J: A gene regulatory program in human breast cancer. Genetics. 201:1341–1348. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Roberts MS, Sahni JM, Schrock MS, Piemonte KM, Weber-Bonk KL, Seachrist DD, Avril S, Anstine LJ, Singh S, Sizemore ST, et al: LIN9 and NEK2 are core regulators of mitotic fidelity that can be therapeutically targeted to overcome taxane resistance. Cancer Res. 80:1693–1706. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Boulay G, Awad ME, Riggi N, Archer TC, Iyer S, Boonseng WE, Rossetti NE, Naigles B, Rengarajan S, Volorio A, et al: OTX2 activity at distal regulatory elements shapes the chromatin landscape of group 3 medulloblastoma. Cancer Discov. 7:288–301. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Feng X, Guo J, An G, Wu Y, Liu Z, Meng B, He N, Zhao X, Chen S, Zhu Y, et al: Genetic aberrations and interaction of NEK2 and TP53 accelerate aggressiveness of multiple myeloma. Adv Sci (Weinh). 9:e21044912022. View Article : Google Scholar : PubMed/NCBI | |
|
Deng L, Sun J, Chen X, Liu L and Wu D: Nek2 augments sorafenib resistance by regulating the ubiquitination and localization of β-catenin in hepatocellular carcinoma. J Exp Clin Cancer Res. 38:3162019. View Article : Google Scholar : PubMed/NCBI | |
|
Nabilsi NH, Ryder DJ, Peraza-Penton AC, Poudyal R, Loose DS and Kladde MP: Local depletion of DNA methylation identifies a repressive p53 regulatory region in the NEK2 promoter. J Biol Chem. 288:35940–35951. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Ji YY, Meng M and Miao Y: lncRNA SNHG1 Promotes progression of cervical cancer through miR-195/NEK2 axis. Cancer Manag Res. 12:11423–11433. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Xia B, Zhao Z, Wu Y, Wang Y, Zhao Y and Wang J: Circular RNA circTNPO3 regulates paclitaxel resistance of ovarian cancer cells by miR-1299/NEK2 signaling pathway. Mol Ther Nucleic Acids. 21:780–791. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Fang Y and Zhang X: Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle. 15:895–907. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Naro C, Barbagallo F, Chieffi P, Bourgeois CF, Paronetto MP and Sette C: The centrosomal kinase NEK2 is a novel splicing factor kinase involved in cell survival. Nucleic Acids Res. 42:3218–3227. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z, et al: NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 23:48–62. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Neal CP, Fry AM, Moreman C, McGregor A, Garcea G, Berry DP and Manson MM: Overexpression of the Nek2 kinase in colorectal cancer correlates with beta-catenin relocalization and shortened cancer-specific survival. J Surg Oncol. 110:828–838. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Lim J, Hwang YS, Kim JT, Yoon HR, Park HM, Han J, Kwon T, Lee KH, Cho HJ and Lee HG: NEK2 phosphorylates RhoGDI1 to promote cell proliferation, migration and invasion through the activation of RhoA and Rac1 in colon cancer cells. Cells. 13:20722024. View Article : Google Scholar : PubMed/NCBI | |
|
Takahashi Y, Iwaya T, Sawada G, Kurashige J, Matsumura T, Uchi R, Ueo H, Takano Y, Eguchi H, Sudo T, et al: Up-regulation of NEK2 by microRNA-128 methylation is associated with poor prognosis in colorectal cancer. Ann Surg Oncol. 21:205–212. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Yi YW, You KS, Park JS, Lee SG and Seong YS: Ribosomal protein S6: A potential therapeutic target against cancer? Int J Mol Sci. 23:482021. View Article : Google Scholar : PubMed/NCBI | |
|
Grasso S, Tristante E, Saceda M, Carbonell P, Mayor-López L, Carballo-Santana M, Carrasco-García E, Rocamora-Reverte L, García-Morales P, Carballo F, et al: Resistance to Selumetinib (AZD6244) in colorectal cancer cell lines is mediated by p70S6K and RPS6 activation. Neoplasia. 16:845–860. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Wan H, Xu L, Zhang H, Wu F, Zeng W and Li T: High expression of NEK2 promotes gastric cancer progression via activating AKT signaling. J Physiol Biochem. 77:25–34. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Chang YY, Yen CJ, Chan SH, Chou YW, Lee YP, Bao CY, Huang CJ and Huang W: NEK2 promotes hepatoma metastasis and serves as biomarker for high recurrence risk after hepatic resection. Ann Hepatol. 17:843–856. 2018. View Article : Google Scholar : PubMed/NCBI |










